
Translate this page into:
A pharmacokinetic profiling study after single-tablet regimen of camylofin 50 mg and paracetamol 325 mg in healthy participants
*Corresponding author: Krishnapriya Mohanraj, Department of Pharmaceutical Analysis, Bombay College of Pharmacy, Mumbai, Maharashtra, India. krishnapriyamohanraj@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Kudyar P, Raj JP, Kulkarni AA, Raju AP, Mallayasamy S, Mohanraj K. A pharmacokinetic profiling study after single-tablet regimen of camylofin 50 mg and paracetamol 325 mg in healthy participants. Indian J Physiol Pharmacol. 2024;68:183-8. doi: 10.25259/IJPP_86_2024
Abstract
Objectives:
Camylofin is widely used in combination with non-steroidal anti-inflammatory drugs such as paracetamol and nimesulide, but there is a dearth of information about its pharmacokinetic properties. Here, we assessed the pharmacokinetic parameters of a fixed-dose combination of camylofin 50 mg and paracetamol 325 mg in healthy volunteers.
Materials and Methods:
Eligible participants were admitted for fasting and fed visits, where 5 mL of blood was collected at multiple time points over 8 h. Serum concentrations of both drugs were analysed using the method of liquid chromatography/tandem mass spectrometry. Key primary pharmacokinetic outcome measures that were analysed were maximum concentration (Cmax), time to maximum concentration (tmax), area under the curve (AUC0-8hrs), elimination constant (Kel), volume of distribution (Vd), clearance (CL) and half-life (t1/2).
Results:
Pharmacokinetic analysis was performed for 12 enrolled participants in both fasting and fed states. Mean (standard deviation [SD]) t1/2 (h) of camylofin was 1.16 (0.53) and 1.68 (0.55) in the states of fasting and fed, respectively, of paracetamol was 2.3 (0.35) in both states. Mean (SD) absolute Vd (L) was 32123.3 (15630.9) and 32928 (14734.4) for camylofin and 93.27 (31.73) and 87.61 (15.48) for paracetamol, respectively. Mean (SD) CL (L/hr) in the two states was 22958.33 (14314) and 14213 (7433.46) for camylofin and 28.07 (7.3) and 26.68 (4.86) for paracetamol, respectively. Mean (SD) Kel was 0.69 (0.26) and 0.46 (0.17) for camylofin, 0.31 (0.04), and 0.30 (0.04) for paracetamol, respectively.
Conclusion:
In the absence of other studies on pharmacokinetic parameters, findings from the current study may be considered as a reference for future research and development on camylofin.
Keywords
Camylofin
Paracetamol
Pharmacokinetic parameters

INTRODUCTION
Camylofin has been indicated for symptomatic relief in gastrointestinal, renal or ureteric colic, menstrual colic or primary dysmenorrhea.[1] It hastens the process of cervical dilatation for augmentation of labour, providing enhanced analgesia along with drugs like tramadol.[2-4] It has been available in India since the year 1958 and has traversed almost six decades of clinical use.[5] The antispasmodic properties of camylofin were first described by Brock in 1951.[6] It is a phosphodiesterase type intravenous (IV) inhibitor having mild calcium-channel blocking effects structurally being related to papaverine, which directly acts on smooth muscle cells, acting as a spasm inhibitor.[7] It is available as an intramuscular injectable as well as in the oral form; it has been orally marketed as fixed-dose combinations (FDCs) along with non-steroidal anti-inflammatory drugs (NSAIDs) such as paracetamol (also called acetaminophen), nimesulide, diclofenac and mefenamic acid.
Sufficient information is already available on the structural, physical and chemical properties of camylofin, and several studies have also emphasised the efficacy and safety of the drug[8-10], but there was a dearth of information regarding the pharmacokinetic parameters of camylofin. Therefore, this study was proposed with the objective of assessing the pharmacokinetic profile of camylofin and paracetamol after a single dose of an FDC of camylofin and paracetamol in normal healthy individuals both in fed and fasting states.
MATERIALS AND METHODS
Ethics
The study was done in accordance with the principles of Good Clinical Practice, the Declaration of Helsinki, the National Guidelines for Ethical Research in Human Participants (Indian Council of Medical Research Guidelines, 2017) and the new drugs and clinical trials rules (Central Drugs Standard Control Organisation, 2019). The Institutional Ethics Committee approval was taken before starting the study and it was prospectively registered in the Clinical Trials Registry of India (2021/03/032174). A written informed consent was taken from all the potential participants after they agreed to participate in the study.
Study design and study setting
It was a non-randomised, open-label, single-dose and crossover (fed and fasted states) study conducted from 09th to 26th May 2022 at a single tertiary care teaching hospital in Mumbai.
Recruitment strategy and eligibility criteria
Healthy participants were identified from among relatives of patients who were visiting the Outpatient Department of our institute.
Adults of any sex, 18 through 60 years of age, who were willing to provide written informed consent and were healthy, as determined by the medical history and clinical and laboratory evaluation, were included in the study. Those with abnormal laboratory findings, those unwilling to perform a coronavirus disease (COVID) screening test, and those in whom establishing IV access was difficult were excluded from the study.
Since we did not have the PK data to estimate the sample size using one of the statistical methods, we followed the recommendation from best practices in bioequivalence and bioavailability studies, which are influenced by both regulatory expectations and empirical research. For instance, the european medicines agency states that ‘The number of evaluable subjects in a bioequivalence study should not be <12’ (Clause 4.1.3).[11]
Study intervention
A single dose of camylofin 50 mg with paracetamol 325 mg (Bigspas-P™) FDC was given to the participants with 240 mL of water on each of the dosing visits.
Study procedures
The research team members counselled the participants, and subsequently, written informed consent was obtained before the screening. On the same day, a thorough medical history, including a history of COVID-19-related symptoms, and a general and systemic examination were done. Subsequently, 15 mL of blood was collected to screen for baseline laboratory investigations.
Eligible healthy participants were requested to get admitted after a 10-hour fasting period on a mutually convenient day. Each participant also had a peripheral vein catheter of wide bore, and this was used in the collection. The phlebotomists were trained, and several dry runs were conducted on study investigators so that their phlebotomy skills with respect to this study were standardised. A baseline (0 h) blood sample was taken, and the study medication was administered. Post this, samples were collected at the following time points: 0.25 h (±3 min), 0.5 h (±3 min), 0.75 h (±3 min), 1.0 h (±3 min), 1.25 h (±3 min), 1.5 h (±3 min), 1.75 h (±3 min), 2 h (±10 min), 3 h (±10 min), 5 h (±10 min) and 8 h (±10 min). A total of 12 time points were planned on the dosing day, with 5 mL of blood withdrawn at each time point, amounting to not more than 60 mL in total. The time points of each patient were monitored by the study team using digital synchronised clocks to avoid any deviations. A standard lunch was provided by the study investigators to the participants at 4 h post the dosing.
The second visit was planned for at least 2 days but within 7 days after the first visit to ensure complete washout of the drug, which was assumed based on the half-life of paracetamol. Similar procedures were followed, except that the study drug was administered after a standard breakfast provided by the study team. The study flow diagram is depicted in Supplementary Figure 1.
Biological sample handling
Each blood sample was centrifuged at a speed of 2500 rpm within 1 h of blood collection, and the sera were stored in a − 20°C deep freezer for further processing. Analysis of both camylofin and paracetamol concentrations was performed as batches using a pre-specified protocol on a liquid chromatography-tandem mass spectrometry (LC-MS/MS) instrument. Several dry runs before the start of the study and the quality control measures during the study conduct were in place.
Method of analysis
An Agilent 1260 series liquid chromatography system attached to Sciex 5500 QTrap tandem mass spectrometer (LC-MS/MS) equipped with a quaternary pump, autosampler and a column oven along with a reverse-phase high-performance liquid chromatography column having an octadecylsilane stationary phase was used for the study. Flow injection analysis technique was used to optimise LC-MS/MS conditions such as GS1 flow, GS2 flow, temperature and the like. The developed method (unpublished data) was validated as per the US FDA Centre for Drug Evaluation and Research guidelines for bioanalytical method validation.
Post-validation, the serum samples obtained from the study participants were spiked with internal standard, and a protein precipitating agent was added. The solution was vortexed and centrifuged to obtain a clear supernatant. The clear supernatant obtained was injected into the LC-MS/MS system using a valco valve to vent out the serum components before the analytes entered the MS/MS. A calibration curve for paracetamol consisting of nine standard solutions and 11 for camylofin spiked in the blank serum was subjected to a similar sample treatment, and the concentration of paracetamol and camylofin was estimated against the standard calibration curve.
Study outcome measures
Pharmacokinetic parameters such as maximum concentration (Cmax), time required to achieve maximum concentration (tmax), area under the curve till time t (AUC0-t), AUC till infinity (AUC0-∞), elimination constant (Kel), volume of distribution (Vd), clearance (CL) and half-life (t1/2) were estimated for both camylofin and paracetamol in the fasted and fed states.
Data management and quality assurance
All the relevant data of each participant were captured in a case record form that was specially designed for the study. All the data entry was done in Microsoft Excel (Publisher: Microsoft Corporation, Redmond, Washington, USA, 2016). Internal monitors independent of the study performed a 100% source data verification and assessed if the study was conducted in concurrence with the approved protocol as per the ethics and regulatory guidelines without any violation of the rights and safety of the participants.
Statistical and pharmacokinetic analysis
No formal sample size estimation was made for this study as no preliminary data were available on the pharmacokinetics of camylofin. Hence, 12 normal healthy participants were recruited in the study. Descriptive statistics such as mean, standard deviation (SD), median, interquartile range and coefficient of variation were used to describe the various pharmacokinetic parameters. Data visualisation was performed with R software v4.2.1[12] using ggplot2 v3.3.6. [13] Non-compartmental analysis was done for the pharmacokinetic parameters using Julia computing language v1.7.1 with the Pumas package.[14]
RESULTS
Subject disposition
In all, 19 healthy volunteers were screened, of which 12 were enrolled. Subject disposition is shown in Figure 1. All recruited participants completed both visits as per the protocol.
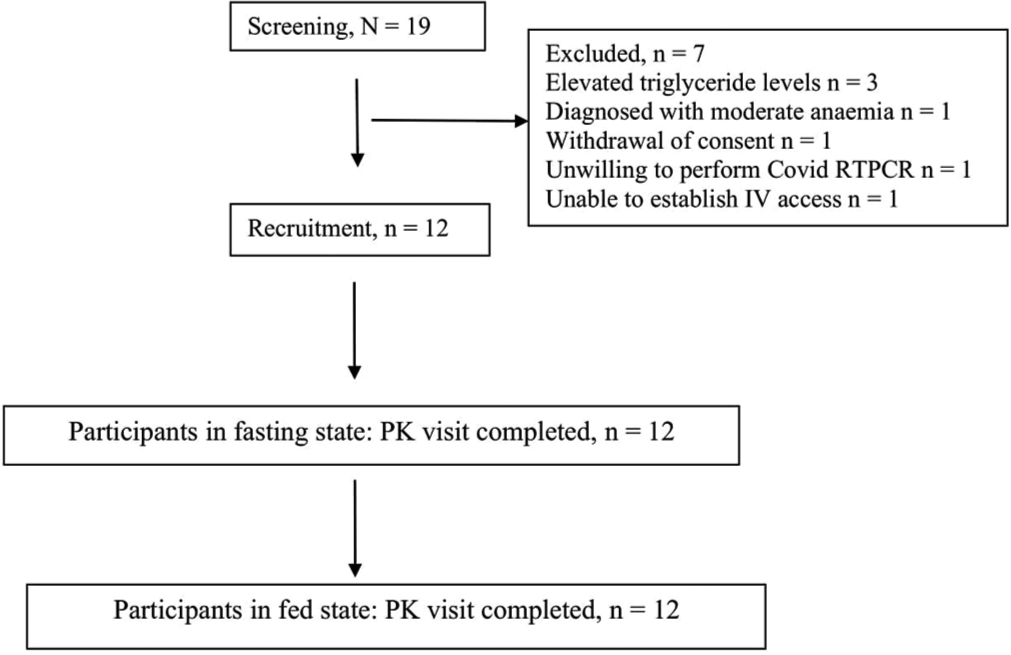
- Participant disposition. RTPCR: Reverse transcription polymerase chain reaction; PK: Pharmacokinetics
Baseline characteristics
The mean (SD) age of study participants was 39.33 (6.72) years, and the minimum age was 21, whereas the maximum age was 46. Their mean (SD) body mass index was 23.1 (1.82) kg/m2. A total of 91% (n = 11/12) of the participants were male. The baseline demographic characteristics are shown in Table 1, and the baseline laboratory parameters are summarised in Supplementary Table 1.
| Characteristics | Number of participants (n=12) |
|---|---|
| Age (years) | |
| 18–30 | 1 |
| 31–45 | 10 |
| 46–59 | 1 |
| Sex | |
| Males | 11 |
| Females | 1 |
| BMI (kg/m2) | |
| <18.5 | 0 |
| 18.5–24.9 | 12 |
| ≥25 | 0 |
BMI: Body mass index
Pharmacokinetic analysis of camylofin and paracetamol
Kel and parameters depending on Kel could not be estimated for camylofin in two participants and for paracetamol in five participants in the fed state as there was a lack of data post the maximum concentration for these participants.
The mean (SD) Cmax of camylofin was 2.27 (1.67) ng/mL and 2.43 (2.63) ng/mL, and that of paracetamol was 4646.67 (1679.02) ng/mL, and 3538.57 (858.04) ng/mL in the states of fasting and fed, respectively. The serum concentrations of both camylofin and paracetamol were lower in fed states than in fasting states in the absorption phases of the drugs, as shown in superimposed plots in Figure 2.
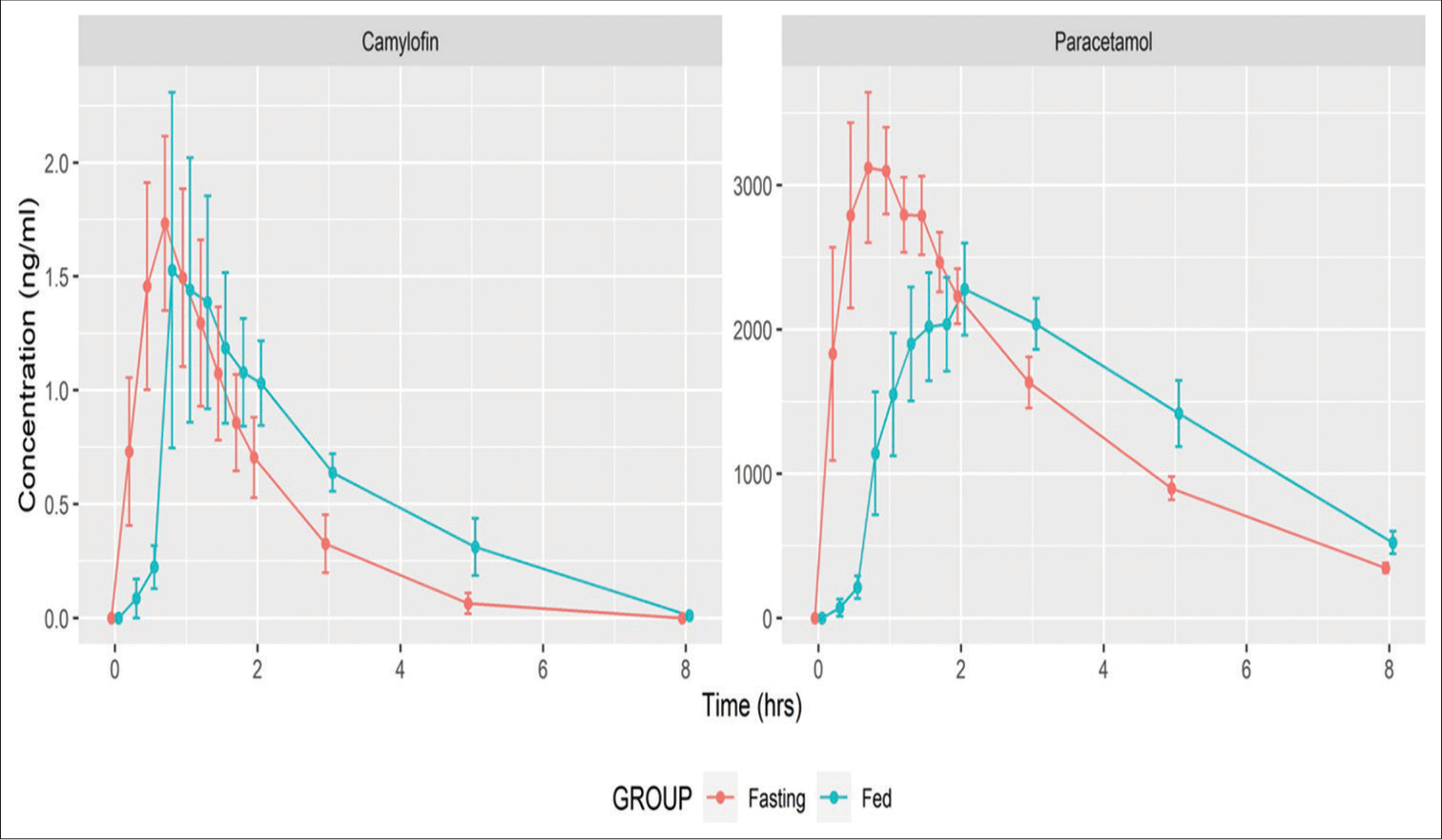
- Mean serum concentration analysis of individual drugs in fasting and fed states.
The mean (SD) Tmax of camylofin was 0.83 (0.34) and 1.05 (0.47) h, while that for paracetamol was 0.854 (0.457) and 1.357 (0.517) h in the states of fasting and fed, respectively. The mean (SD) half-life of camylofin was 1.16 (0.53) h and 1.68 (0.55) h in fasting and fed states, respectively, whereas the half-life of paracetamol was 2.3 (0.35) h in both fasting and fed states. The mean (SD) absolute CL for camylofin was 22958.33 (14314) L/h and 14213 (7433.46) L/h, while for paracetamol, it was 28.07 (7.3) L/h and 26.68 (4.86) L/h in fasting and fed states, respectively. A summary of all pharmacokinetic parameters is presented in Table 2.
| Parameter, mean (SD) | Camylofin | Paracetamol | ||
|---|---|---|---|---|
| Fasting state | Fed state | Fasting state | Fed state | |
| AUC0-∞ (ng h/mL) | 3.625 (2.948) | 4.624 (2.976) | 12485 (3976.352) | 12531.428 (2299.764) |
| AUC0-8hrs (ng h/mL) | 2.905 (2.74) | 3.724 (3.008) | 11324.166 (3761.36) | 11041.428 (1992.489) |
| Absolute clearance (L/h) | 22958.3 (14314) | 14213 (7433.46) | 28.066 (7.304) | 26.685 (4.858) |
| C max (ng/mL) | 2.265 (1.669) | 2.431 (2.632) | 4646.67 (1679.022) | 3538.57 (858.049) |
| Half-life (h) | 1.16 (0.535) | 1.681 (0.551) | 2.280 (0.356) | 2.298 (0.351) |
| Kel | 0.696 (0.256) | 0.46 (0.168) | 0.310 (0.044) | 0.307 (0.045) |
| Tmax (h) | 0.835 (0.344) | 1.05 (0.468) | 0.854 (0.457) | 1.357 (0.517) |
| Absolute Vd (L) | 32123.3 (15630.9) | 32928 (14734.4) | 93.266 (31.733) | 87.614 (15.479) |
SD: standard seviation, AUC: area under the curve
Safety
No adverse physical symptoms such as drowsiness, headache, dry mouth, nausea, vomiting, or constipation were reported by any study participants. There were no drug-related events that resulted in the withdrawal of the study treatment, withholding of study treatment, reduction of the dose of the study drug, or requirement for the use of any additional concomitant treatment.
DISCUSSION
The present study evaluated the pharmacokinetic parameters of FDCs of camylofin 50 mg and paracetamol 325 mg in the states of both fasting and fed in a total of 12 participants with a mean (SD) age of study participants 39.33 (6.7) years. The mean concentration of camylofin was obtained in nanograms and that of paracetamol was obtained in micrograms.
The levels of serum concentration of camylofin were very low (nanograms), whereas the Vd was very high in ranges above 103 L. This suggests that camylofin is likely to be tissue-bound and that it gets distributed widely, resulting in a low concentration in the serum.[15] Furthermore, camylofin is likely more lipid soluble. This could be explained by the fact that the esterase enzyme presents in the body cleaves the camylofin molecule into two pharmacologically weak acidic metabolites, which are isoamyl alcohol and alpha-N-(beta-diethyl aminoethyl) amino-phenylacetic acid,[16] though these were not estimated in our study. It is known that weakly acidic drugs are more lipid soluble, which we hypothesise will facilitate the crossing of camylofin across the cell membranes. Lipid solubility will also increase the apparent space available for dilution.[17] as well as CL in both fasting and fed states.[18] This is corroborated by the fact that in the fed state, such as any other lipid-soluble drug, the concentration of the drug was lower in the absorption phase than in the fasting state.
Greater lipid solubility would also facilitate greater penetration across the blood-brain barrier, and it is likely that it also exerts a central effect by acting on the central nervous system like the other NSAIDs.[19] This attribute of camylofin could explain the efficacy of camylofin despite a low serum concentration. It is also well known that food affects drug absorption by the mechanisms of delaying gastric emptying time, altering the gastrointestinal pH, stimulating the bile flow, increasing splanchnic blood flow or by the process of physically interacting with drugs.[20,21] This is reflected by the fact that in the fed state, the Tmax is delayed for the drug.
Paracetamol is a widely used analgesic and antipyretic agent that is well absorbed in the gastrointestinal tract. It is an extremely weak acid essentially unionised at physiological pH values with its binding to plasma proteins being negligible.[22] The free (unbound) drug concentrations that are present in the plasma are known to make the drug more readily available to transfer to tissues as well as for extravasation and crossing cell membranes, which is reflected by the high serum concentrations of the drug, as seen in our analysis.[23] The average CL of the drug is 20 L/h with a half-life of 2.5 h, which is comparable to the pharmacokinetic parameters obtained in our study.[24] High carbohydrate food is known to decrease paracetamol peak plasma concentration by up to 4 times, which is reflected in the decreased maximum concentration of the drug in the fed state. Even in the state of fasting, the rate of absorption of paracetamol is mostly variable, with the maximum plasma concentration being reached after 20 min up to 1.5 h.[25] This is like the results of our study where the time required to achieve maximum concentration increased from fasting to fed state, with a mean of 51 min in the fasting state.
Our study was the first of its kind study to elaborate on the pharmacokinetic parameters of camylofin in healthy human participants. The serial blood collections of participants over 8 h in both fasting and fed states gave an elaborate pharmacokinetic profile of both drugs. This will help to establish the dose-response curve and to optimise the drug therapy in its future applications. The study was limited by the fact that it had a small sample size and recommended that larger studies be planned to confirm our study findings and for better generalizability. It was also not a randomised study, and therefore, no formal comparisons were made in the parameters between the states of fasting and feeding.
CONCLUSION
With the administration of camylofin-paracetamol FDC at doses of 50 mg and 325 mg, respectively, in 12 participants, it was seen that the serum concentration of camylofin was quite low, being in nanograms, and for paracetamol concentrations were in micrograms. The pharmacokinetic parameters of paracetamol were comparable to those reported in other studies. It was also observed that, in most cases, in the fasting state, both Cmax and Tmax for camylofin were lesser than that in the fed state, and for paracetamol, Cmax was higher in the fasting state, and the Tmax was higher in the fed state, though, this was not statistically evaluated in this study. The Vd and CL of camylofin were very high. Thus, in the absence of other studies to compare the pharmacokinetic parameters of camylofin, the present study may be considered as a reference study for future research and development of camylofin.
Ethical approval
The research/study was approved by the Institutional Review Board at Seth GS Medical College and KEM Hospital, Mumbai, number EC-OA-120/2020, dated 12 February 2021.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Supplementary data available at:
Financial support and sponsorship
Abbott Healthcare Pvt Ltd.
References
- Role of camylofin and its combinations in obstetrics and gynaecological practice: A review of Indian evidence. Int J Reprod Contracept Obstet Gynecol. 2019;8:359-67.
- [CrossRef] [Google Scholar]
- Camylofin in the management of prolonged labor: A review of evidence. Int J Reprod Contracept Obstet Gynecol. 2017;6:776-80.
- [CrossRef] [Google Scholar]
- Programmed labor: Indegenous protocol to optimize labor outcome. South Asian Fed Obstet Gynecol. 2009;1:61-4.
- [CrossRef] [Google Scholar]
- Active management labor in a low-resource setting and its impact on cesarean section rates. Int J Gynecol Obstet. 2006;94:54-5.
- [CrossRef] [Google Scholar]
- Unpublished Data: The drugs controller, India - letter of permission to import 'Avacan' Mumbai: Courtesy-Abbott Healthcare Private Limited; 1958.
- [Google Scholar]
- Effectiveness and safety of camylofin in augmentation of labor: A systematic review and meta-analysis. J Obstet Gynaecol India. 2020;70:425-39.
- [CrossRef] [Google Scholar]
- A randomized comparative study of intramuscular camylofin dihydrochloride and intravenous drotaverine hydrochloride on cervical dilatation in labor. Indian J Clin Pract. 2015;26:157-62.
- [Google Scholar]
- The efficacy of camylofin dihydrochlorid in acceleration of labour. A randomized double-blind trial. J Bombay Hosp. 2003;45:420-4.
- [Google Scholar]
- Guidelines for BA/BE. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf [Last accessed on 2024 May 27]
- [Google Scholar]
- R: A language and environment for statistical computing. 2022. Vienna, Austria: R Foundation for Statistical Computing; Available from: https://www.R-project.org [Last accessed on 2023 Sep 22]
- [Google Scholar]
- Ggplot2. In: Elegant graphics for data analysis. New York: Springer-Verlag; 2016.
- [CrossRef] [Google Scholar]
- Accelerated predictive healthcare analytics with pumas, a high performance pharmaceutical modelling and simulation platform. BioRxiv; 2020
- [CrossRef] [Google Scholar]
- Drug distribution to tissues. Available from: https://www.msdmanuals.com/en-in/professional/clinical-pharmacology/pharmacokinetics/drug-distribution-to-tissues [Last accessed on 2023 Nov 16]
- [Google Scholar]
- Comparative efficacy of camylofin dihydrochloride and drotaverine hydrochloride on cervical dilatation in active labour. Int J Clin Obstet Gynaecol. 2021;5:294-7.
- [CrossRef] [Google Scholar]
- Beyond the blood: Brain barrier: The importance of central nervous system (CNS) pharmacokinetics for the treatment of CNS tumors, including diffuse intrinsic pontine glioma. Front Oncol. 2018;8:239.
- [CrossRef] [Google Scholar]
- Plasma drug concentrations: Description and interpretation of the biexponential decay. Br J Anaesth. 1993;71:908-14.
- [CrossRef] [Google Scholar]
- Drug bioavailability: Estimation of solubility, permeability, absorption and bioavailability. Weinheim: Wiley-VCH GmbH and Co 2009:523-58.
- [CrossRef] [Google Scholar]
- Clinical relevance of drug binding to plasma proteins. J Mol Struct. 2014;1077:4-13.
- [CrossRef] [Google Scholar]
- Paracetamol pharmacokinetic parameters. Available from: https://sepia2.unil.ch/pharmacology/drugs/paracetamol [Last accessed on 2023 Mar 16]
- [Google Scholar]
- The modern pharmacology of paracetamol: Therapeutic actions, mechanism of action, metabolism, toxicity, and recent pharmacological findings. Inflammopharmacology. 2013;21:201-32.
- [CrossRef] [Google Scholar]




