
Translate this page into:
Transcranial magnetic stimulation in exploring neurophysiology of cortical circuits and potential clinical implications
*Corresponding author: Kaviraja Udupa, Department of Neurophysiology, National Institute of Mental Health and Neuro-Sciences, Bengaluru - 560 029, Karnataka, India. kaviudupa@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Udupa K. Transcranial magnetic stimulation in exploring neurophysiology of cortical circuits and potential clinical implications. Indian J Physiol Pharmacol 2020;64(4):244-57.
Abstract
Transcranial magnetic stimulation (TMS) is a non-invasive, painless technique to stimulate the human brain. Although it has been used in clinical research both as an investigative tool and treatment modality for the past three decades, its use has been restricted to tertiary health centres or higher-end academic research institutions. The aim of this review is to popularise the concepts of this effective non-invasive brain stimulation technique, further facilitating its use both in research and clinical practice among clinical physiologists. In the first part of this article, a brief physiologic overview of TMS will be provided with basic as well as the basic technical details. This is followed by a discussion of TMS parameters that can be studied using single and paired pulses of TMS which could be used to investigate the altered excitability of cortical circuits. Finally, how rTMS and patterned TMS could be used to induce plasticity which, in turn, could be potentially used as therapeutic interventions in various neurological and psychiatric disorders will be illustrated. In each section of this article, diagnostic as well as therapeutic utilities of TMS in Neurology and Psychiatric disorders will be discussed. These discussions could not only facilitate the understanding of pathophysiology of mood and movement disorders but also to manage various neurological and psychiatric disorders with novel therapeutic options. In the end, few future directions, limitations of this technique and comparison with other techniques will be provided. I hopefully, this review would elicit some interest in physiologists to take up this exciting area of brain stimulation as a research subject and work further on understanding the functions of brain and use it effectively in the management of various brain-related disorders.
Keywords
Transcranial magnetic stimulation
Non-invasive brain stimulation
Mood disorders
Movement disorders
Plasticity

INTRODUCTION AND HISTORY OF TRANSCRANIAL MAGNETIC STIMULATION (TMS)
Since its invention by Barker et al. in 1985,[1] TMS has been used to study the complex circuitries and mechanisms in the brain and as a possible treatment in variety of neurological and psychiatric disorders.[2-6] TMS functions by passing a large (500–4000 V) but brief (around 100 micro seconds) electrical current through a wire coil which is placed over the region to be stimulated (brain, spinal cord, or peripheral nerves), [Figure 1]. The transient current produces a large and changing magnetic field (peak of 1.5–2 Tesla), with lines of flux passing perpendicularly to the plane of the coil. Thus, TMS is based on the principle that a changing electric current in a wire coil induces a changing magnetic field (Ampere’s law) perpendicular to the current flow in the coil.[3] This changing magnetic field passes unimpeded through the scalp and skull and induces an electric current in the brain that flows in the opposite direction to the current in the coil (Faraday’s law), [Figure 2]. The TMS apparatus mainly consists of boosters, control panel connected to a computer and a stimulating coil. Boosters are made up of a capacitor discharge system which works with voltages of 500–4000 V and stores energy in the range of 400–2000 J. Stimulating coil is made up of wire loops encased in insulated plastic and connected through a cable to one or more capacitors. Electrical current passes through the coil for a brief period of 100–200 μs.[3] When the current flows through the coil, brief magnetic field in the range of 1.5–2 teslas is going to be generated depending on the stimulator type, make and machine output selected for a given stimulation protocol.
![Diagrammatic representation of pathways of transcranial magnetic stimulation stimulation over contralateral motor cortex and conduction along the corticospinal tract to evoke electromyography response in terms of motor evoked potential. [Adapted and modified from Klomjai et al. Annals of Physical and Rehabilitation Medicine 58 (2015) 208–213 and Sack AT, Linden DE. Brain Res Brain Res Rev. 2003 Sep;43(1):41-56].](/content/114/2020/64/4/img/IJPP-64-244-g001.png)
- Diagrammatic representation of pathways of transcranial magnetic stimulation stimulation over contralateral motor cortex and conduction along the corticospinal tract to evoke electromyography response in terms of motor evoked potential. [Adapted and modified from Klomjai et al. Annals of Physical and Rehabilitation Medicine 58 (2015) 208–213 and Sack AT, Linden DE. Brain Res Brain Res Rev. 2003 Sep;43(1):41-56].
![Diagrammatic representation of two commonly used types of transcranial magnetic stimulation coils (circular and figure of 8) and the modeling strength of induced current beneath the scalp. [Adapted and modified from Ge S et al, International Scholarly and Scientific Research & Innovation 6(5) 2012 and Cohen et al., Electroencephalogr Clin Neurophysiol, 75 (1990) 350-7].](/content/114/2020/64/4/img/IJPP-64-244-g002.png)
- Diagrammatic representation of two commonly used types of transcranial magnetic stimulation coils (circular and figure of 8) and the modeling strength of induced current beneath the scalp. [Adapted and modified from Ge S et al, International Scholarly and Scientific Research & Innovation 6(5) 2012 and Cohen et al., Electroencephalogr Clin Neurophysiol, 75 (1990) 350-7].
The strength of the induced current is a function of rate of change of the magnetic field, which, in turn, depends on the rate of change of current in the coil. Magnetic field penetrates the scalp and skull much more easily than electrical energy. The induced electrical current depolarises axons, most likely at sites of bending [Figure 1]. The effects of TMS depend on the intensity of the induced magnetic fields, the shape and orientation of the induced current. Some coils, such as a circular coil, stimulate a large brain area. Other coil designs, such as a figure-of-eight coil, produce more focal stimulation with the maximum stimulation at the junction of the two loops [Figure 2]. Different current directions activate different groups of cortical neurons.
MEASURES DERIVED FROM OF SINGLE PULSE TMS
The following parameters employ single pulse of TMS and measures the different aspects of cortical excitability. These measures are: (a) Motor threshold (MT: Minimal stimulation intensity to generate a small motor evoked potential [MEP], Figure 1); (b) MEP amplitude at a given stimulation intensity and recruitment curve (RC, input-output curve measuring MEP amplitudes with increasing stimulation intensities); (c) central motor conduction time (CMCT, time required for the stimulus to conduct through the central part of corticospinal pathways, that is, primary motor cortex to alpha motor neuron in the spinal cord), (d) silent period (SP, a pause in the electromyography activity during voluntary contraction following TMS) and (e) mapping of muscle representation. These measures have been used as diagnostic and prognostic parameters as well as to investigate the neurobiology in various neurological and psychiatric disorders [Table 1]. All the above-mentioned measures such as MT, RC, CMCT, SP and motor cortical mapping quantify the excitability of corticospinal system. Thus, these single TMS pulse measures could provide useful information about the physiology of cortical circuits in patients with neuropsychiatric disorders. Studies have shown that these measurements frequently disclose abnormal findings, but are not specific for any disease per se. Thus, TMS has a role in demonstrating CMCT abnormalities in patients with various neurological and psychiatric disorders in whom pyramidal signs may be clinically equivocal. Hence, TMS proved to be a very useful research tool to investigate the pathophysiology of various neuropsychiatric disorders.
| Disorder | MT | CMCT | SP | MEP amplitude | Recruitment curve | Mapping of muscle representation |
|---|---|---|---|---|---|---|
| PD | ↓/↔ | ↔ | ↓ | ↓ | ↓ | Anterior displacement of hand muscle area |
| Dystonia | ↑/↔ | ↔ | ↓ | ↓ | - | Distorted hand muscle in FHD |
| Huntington’s disease | ↔ | ↔ | C | ↔ | - | - |
| Wilson’s disease | ↑ | ↓ | ↓ | ↑ | - | - |
| Ataxia | ↓ | ↑ | - | ↓ | - | - |
| Major Depression | ↑ | ↔ | ↔ | ↑/↓ | - | - |
| Bipolar disorders | ↓ | ↔ | ↔ | - | - | - |
| Schizophrenia | ↔ | ↑/↓ | ↓ | - | - |
TMS measures in movement disorders (adapted and modified from Ref 91). ↔: normal; ↓: decreased;: increased; C: Controversial/not established; -: not been studied; FHD: Focal hand dystonia. These findings represent results from multiple studies. If studies showed conflicting results, the findings of the majority of studies are shown here. MT: Motor threshold, CMCT: Central motor conduction time, SP: Silent period, MEP: Motor evoked potential, PD: Parkinson’s disease
MT
MT is considered a measure of corticospinal excitability. MT refers to the lowest TMS intensity capable of eliciting small MEPs and is usually defined as more than 50 μV in amplitude in muscles at rest or 200 μV in active muscles in at least five out of ten trials.[4] Thus depending on the state of activity of the muscle group and related corticospinal neuronal pool, MT is classified accordingly as resting (RMT) and active (AMT). Since while performing active muscle contraction, the neuronal excitability is higher than the resting state, AMT values generally lower than RMT.[4] MT likely reflects the membrane excitability of cortical neurons as it is increased by drugs that block voltage-gated sodium channels. Some studies found increased excitability in Parkinson’s disease (PD) reduced MT compared to healthy controls[7,8] but majority of studies show no change as this measure of excitability which depends on various factors such as the disease stage, dopamine medication status (ON or OFF dopaminergic state) and deep brain stimulation state (ON and OFF stimulation). One of the explanations for increased motor excitability in PD is the inability to completely relax the muscles due to tremor and rigidity, which was partially corrected with medications.[5] A study found that resting MT of upper extremity negatively correlates with the Unified PD rating scale (UPDRS) part-III score, suggesting that this measure could reflect the severity of PD.[9] Patients with spinocerebellar ataxia[10] and Friedreich ataxia[11] have reduced cortical excitability compared to healthy controls. On the other hand, in conditions such as dystonia[12] and Wilson’s disease[13] MT is known to increase indicating either decrease in excitability or deficient inhibitory control mechanisms at the motor cortex. Some studies found increased excitability of motor cortex in major depression,[14,15] which is modulated by rTMS therapy on dorsolateral prefrontal cortex (DLPFC) and can be used as predictor of efficacy of such therapy.[16] In schizophrenia, reversal of normal pattern of interhemispheric corticospinal excitability has been observed.[17] In this study, schizophrenics showed 5–10% higher threshold on the right hemisphere than the left whereas healthy control subjects had a nearly 10% higher threshold for the left than the right hemisphere. However, no difference in threshold was observed in schizophrenics compared to controls,[18] though this study examined only unilateral excitability. In patients with chronic cocaine usage, higher resting MTs have been observed.[19] Hence, further studies need to explore the pathophysiology of cortical excitability in psychiatric disorders.
CMCT
CMCT is the conduction time of corticospinal fibres from the motor cortex to motor neurons in the spinal cord or brainstem. It is calculated by subtracting the peripheral conduction time from the latency of the MEP elicited by motor cortical TMS.[20] On stimulating a motor nerve, the M wave is the direct muscle response and the F wave is the muscle response produced by activation of the alpha motoneuron by the antidromic volley. Thus, peripheral conduction time may be calculated using the formula (F wave latency + M wave latency – 1)/2. Other methods of obtaining the peripheral conduction time include electrical or magnetic stimulation over the spine.[20] CMCT in active muscles were found to be normal[21-29] in PD. However, some studies found decreased CMCT with the target muscle at rest in PD,[26] which is normalised dopaminergic medication.[25,27] This may be related to difficulties with muscle relaxation in PD. On the other hand, increased CMCT have been reported in PD patients[28] with Parkin gene mutations, suggesting that this measure may be used to demarcate patients with Parkin mutation from other early onset PD. Prolonged CMCT also found in patients with parkinsonian syndrome secondary to multiple system atrophy or progressive supranuclear palsy[20] due to corticospinal tract involvement in these disorders. Thus CMCT can be used to differentiate the subgroups of certain types of parkinsonian disorders. Patients with SCA have prolonged CMCT, which was most evident in SCA1 and least in SCA2.[10] Although, compared to healthy volunteers, chronic alcoholics exhibited a significant prolongation of CMCT, no correlation was found between CMCT prolongation and intensity and duration of abuse, presence of peripheral neuropathy, or brain atrophy on imaging.[30] However, there are no studies investigating CMCT in other psychiatric disorders in which motor conduction might be normal.
SP
SP is a pause in on-going voluntary EMG activity produced by TMS.[4] It is elicited by single pulse TMS delivered to the motor hot spot of concerned muscular region over M1 during voluntary contraction of the same muscle of interest (usually the finger muscles). While the first part of the SP is due in part to decreased spinal cord excitability, the latter part is almost exclusively due to cortical inhibition. Prolonged SP suggests hyperactivity whereas shortened SP suggests decreased activity of inhibitory circuits in the motor cortex. This type of inhibition is likely mediated by GABAB receptors as it is increased by GABAB agonist baclofen[31] and GABA reuptake inhibitor tiagabine.[32] SP duration is shortened in PD and this reduction was greater on the more affected side.[33] Further studies have shown that dopaminergic treatment normalises this reduction of SP.[34] A study reported paradoxical lengthening of SP with dopaminergic medications in patients with dyskinesia and both dyskinesia and SP were normalised by pallidal stimulation.[35] Thus, SP may be an indirect measure of clinical severity in PD. SP duration is shortened in major depression and normalised with electroconvulsive therapy (ECT) thus indicating the treatment response to ECT in treatment resistant depression.[36] However, other study showed prolonged SP in patients with depression.[37] Since this study[37] did not follow these patents following therapy, chances of these patients being resistant to antidepressant management could be high. Further, study demonstrated that a single session of yoga practice has shown significant lengthening of the cortical SP in the yoga-group as compared to intermittent walking, thereby demonstrated an enhanced cortical GABA-tone following yoga. Thus, yoga training enhances the inhibitory control of motor cortical circuits.[38] However, prolongation of CSP was also observed in first episode schizophrenics compared to healthy controls and prodromal (at risk of developing the disease) subjects,[39] AD with negative correlation of CSP with that of MMSE scores[40] and after single dose of quetiapine.[41] In addition, a study with schizophrenia patients showed that significant prolongation of CSP of left motor cortex (right FDI) compared to healthy controls reflecting compensatory increased GABABergic transmission induced by hyperactivity of the dopaminergic system which, in turn, could be related to anti-psychotic medications. This study also showed reduced short interval intracortical inhibition (SICI) after stimulation of the right motor cortex was observed in first episode of schizophrenics indicating GABAAergic deficit in this condition.[42] Further, a study demonstrated a reduction in length of the SP in patients with schizophrenia.[18] Hence, it needs further exploration of this neurobiological marker to predict the efficacy of therapy in various neurological and psychiatric conditions.
Recruitment curve
This parameter, also known as input-output or stimulus-response curve, refers to the increase in MEP amplitude with increasing TMS intensity.[4] Compared to MT, this measure assesses neurons that are intrinsically less excitable or spatially further from the centre of activation by TMS. Recruitment curves are likely related to the strength of corticospinal projections and are generally steeper in muscles with low MT, such as intrinsic hand muscles. A study measuring RC in Parkin mutation positive PD patients and gene carriers found no difference in RC among PD patients, gene carriers and controls.[28] In addition, normal RC was also observed in patients with Myoclonus-Dystonia and paroxysmal kinesigenic dyskinesia. However, more studies need to be performed in various disorders to understand this measure of cortical excitability in various other neurological and psychiatric conditions.
Mapping of muscle representation
Mapping is performed by stimulation at a number of different scalp positions with a focal figure-of-eight coil. The number of excitable scalp positions, location of the optimal position for stimulation and the centre of gravity (an amplitude-weighted representative position on the motor map) can be determined. Motor maps are affected by both the location and excitability of the motor representation. A study has shown increased cortical representation of body muscles over the motor strip in PD, probably due to tonic hyperactivation of motor cortical circuitry.[43]
Thus, various single pulse TMS measures are useful in investigating motor cortical excitability of PD [Table 1]. Although many studies investigated excitability changes in PD compared to healthy controls, there is lack of longitudinal studies investigating these single pulse TMS measures comprehensively and following with the course of the disease and including large number of patients to account for the variability of these measures due to many factors such as clinical phenotypes, disease stage and medication profile of the patients.
Paired TMS techniques involve delivering two TMS pulses at different interstimulus intervals (ISI) to study the motor cortical physiology. The first one is usually known as the conditioning stimulus (CS) and the second the test stimulus (TS). Modulation of motor cortical excitability is often calculated as the ratio of MEP amplitude or area produced by CS followed by TS to that of TS alone. Paired TMS techniques are useful ways to assess cortical excitability and physiology of intracortical circuits. The circuits activated depend on the stimulus intensities (different neuronal circuits have different stimulation thresholds), ISI (these circuits have different time constants), area of stimulation and coil orientation (activation of specific neuronal population). The different protocols can be broadly classified as shown in [Figure 2 and Table 2] will be discussed briefly.
| Parameters | SICI | LICI | SICF | ICF | SIHI | LIHI | CBI | SAI | LAI |
|---|---|---|---|---|---|---|---|---|---|
| CS/S1 for SICF) | Sub-threshold TMS | Supra-threshold TMS | Supra-threshold TMS | Sub-threshold TMS | Supra-threshold TMS | Supra-threshold TMS | Cerebellar stim | Median nerve stim | Median nerve stim |
| TS/S2 for SICF | Supra-threshold TMS | Supra-threshold TMS | Sub-threshold TMS | Supra-threshold TMS | Supra-threshold TMS | Supra-threshold TMS | Supra-threshold TMS | Supra-threshold TMS | Supra-threshold TMS |
| Interstimulus interval (ms) | 1–6 | 50–200 | 1.0-1.5, 2.3-3.0, 4.1-5.0 | 8–30 | 8–12 | ~40 | 5-7 | ~20 | ~200 |
| Proposed neurotransmitter/ receptor | GABAA?dopamine | GABAB | ?Glutamate (↓by GABAA) | Glutamate | ? | GABAB | ? | ACh ↑by GABAA | ? |
| Finding in diseases | |||||||||
| PD | ↓ | ↑ | ? | ↓/↔ | ↔ | ↑ | ↓ | ↔ (↓ on meds) | ↓ |
| Dystonia | ↓ | ↑ | ? | ↔ | ↓ | ? | ? | ↔ | ↓ |
| Alzheimer’s disease | ↓ | ? | ↔ | ↓/↔ | ? | ? | ? | ↓ | ? |
| Cerebellar degeneration | ↔ | ↑ | ? | ↓ | ? | ? | ↓ | ? | ? |
| Major depression | ↓ | ↑/↓ | ? | ↓ | ↓ | ↓ | ? | ? | ? |
| Bipolar disorders | ↓ | ↑ | ? | ↔ | ↓ | ↓ | ? | ? | ? |
| Schizophrenia | ↓ | ↓ | ? | ↔ | ↓/delayed | ↓/ delayed | ? | ↓ | ? |
| Tourette’s syndrome | ↓ | ? | ? | ↑ | ? | ? | ? | ↓ | ? |
Ach: acetylcholine, CBI; cerebellar inhibition, ICF: intracortical facilitation, GABA: γ-aminobutyric acid, LICI: Long interval intracortical inhibition, LIHI: long interval interhemispheric inhibition LAI: Long latency afferent inhibition, SAI: short latency afferent inhibition, SICI: short interval intracortical inhibition, SICF: short interval intracortical facilitation, SIHI: short interval interhemispheric inhibition ↓: decreased, ↑: increased, ↔: no change, ?: unknown. PD: Parkinson’s disease, CS: Conditioning stimulus, TS: Test stimulus
Intracortical inhibitory circuits
Short interval intracortical inhibition (SICI)
This paired pulse technique involves a subthreshold CS, followed by a suprathreshold TS with ISI of 1–6 ms.[44] It has been established by recording of corticospinal volleys at cervical epidural space that the inhibition occurs in the motor cortex rather than in spinal level or subcortical structures. The inhibition is biphasic having two phases of maximum inhibition at ISIs of 1 and 2.5 ms. The first phase (1 ms) may partly reflect axonal refractoriness but neuronal inhibition may also contribute. The second phase (2.5 ms) is likely related to cortical inhibition. Moreover, SICI is reduced just before and during voluntary movement contraction. This could serve to ‘release’ cortical representations from inhibition and focus subsequent excitatory drive to produce the intended movement. Although SICI is most extensively studied the hand area of the M1, this inhibitory phenomenon can also be observed in cortical areas representing face, proximal arm and leg. SICI can be enhanced by drugs that increase GABAA activity and by anti-glutaminergic drugs, whereas ion-channel blocking drugs have no effect on these parameters. Thus, SICI may provide information on GABAA and glutaminergic systems in the motor cortex. SICI is reduced in several neurological and psychiatric disorders including PD,[45] dystonia,[46] AD,[47] Tourette’s syndrome[48] and schizophrenia.[42] The reduction in SICI in PD could be due to increased facilitation observed especially during OFF medication[49] rather than the decrease in GABArgic inhibition.[50] In similar lines, patients with rapid eye movement sleep behaviour disorders exhibited a significant decrease of intracortical facilitation (ICF) and trends toward reduction of SICI compared to controls suggesting changes of ICF and, to a lesser extent, SICI (which are largely mediated by glutamatergic and GABAergic transmission, respectively) might precede the onset of a future neurodegeneration. Further, SICI was correlated with the muscle tone alteration, possibly supporting the proposed RBD model of retrograde influence on the cortex from the brainstem.[51] Changes in SICI may also mediate cortical plasticity following amputation or muscle transfer.[52] Studies have shown decreased SICI with depression which recovers with effective antidepressant therapies.[53-56] Depression patients with treatment resistance (failure of two antidepressant therapy regimens consecutively) exhibited reduced SICI and long-interval intracortical inhibition (LICI), as shown by abnormally higher estimates, than those with non-treatment resistance or healthy controls. Further, TRD was characterised by relatively reduced CI, including both GABAA- and GABAB-receptor-mediated neurons while non-TRD preserved partial CI. In non-TRD, SSRIs may mainly modulate GABAB-receptor-related LICI. Thus, these measures could be used as markers of Rx resistance.[36,37,54-57]
SICI has been impaired in patients with schizophrenia and it had significant inverse correlations with emotion processing and a global social-cognition score.[58] Further this reduction of SICI correlated to impaired performance of working memory talk.[59] Thus these measures of intracortical inhibition could be used as prognostic markers or indicators behavioural abnormality. The reduced SICI was normalised by transcranial direct current stimulation (tDCS) and this restoration of the cortical inhibition could be one of the ways by which tDCS improves the condition.[60]
Schizophrenia patients with Cannabis dependence showed greater SICI compared to non-dependent patients[61] indicating differential effects of cannabis dependence on cortical inhibition in schizophrenia patients which could provide insights into the pathophysiology of addiction in schizophrenia. This aspect of dependence has been further worked out in a study and found altered cortical excitability without changes in SICI in patients with alcohol withdrawal syndrome.[62] Further research need to explore the changes in cortical circuits and their relationship to drug dependence and withdrawal.
Even in systemic disorders such as hepatic encephalopathy where GABAergic dysfunctions have been considered, study using investigative TMS have shown flattening of recruitment curve and reduced SICI challenging classical hypothesis of enhanced GABAergic tone in this condition.[63]
Long interval cortical inhibition (LICI)
LICI is evoked by a CS at above threshold intensity followed by a TS at longer ISI of 100–200 ms. LICI is known to be mediated by GABAB mediated receptors which typically acts through metabotropic receptors and have longer latency compared to ionotropic receptors associated with GABAA mediated receptors (seen with SICI).[64] Studies found increased LICI in PD and dystonia subjects patients.[65-67] These GABAergic abnormalities in LICI were normalised with dopaminergic medications in PD[65] and apomorphine.[68] Further, inability of PD patients to completely relax during these investigative sessions might have been an issue, as LICI during muscle activation was found to be increased in PD.[65] Although SP and LICI are thought to be mediated by GABAB, PD tends to affect these two measures in opposite directions. A decrease in SP could be due to reduced inhibition of the corticospinal drive during tonic muscle contraction, whereas an increase in LICI may correspond to reduced motor cortical activity associated with the impaired execution of rapid movements. The opposite findings for SICI and LICI may be because LICI inhibits SICI, likely through presynaptic GABAB receptors.[69]
Such differential involvement of these two circuits is also observed in bipolar disorders, in which SICI is reduced and LICI is increased in patients with medication-naïve/free symptomatic manic and first episode mania when compared to healthy controls[70] demonstrating the disparity of 2 types of GABA receptors involved in these intracortical inhibitory circuits. LICI is impaired in patients with Alzheimer’s disease in spite of therapeutic regimens (monotherapy or combination therapies) compared to healthy controls.[71] In similar lines of SICI, LICI also showed association between increased cortical inhibition and suicidal ideation in adolescent depression patients.[56] Further, LICI computed with TMSEEG techniques showed reduced frontal cortex inhibition in schizophrenia patients compared with healthy controls.[72]
Intracortical facilitation (ICF)
ICF evoked by a CS at below threshold intensity followed by a TS with ISI of 10–20 ms. This TMS measure is found to be either decreased[73] or normal[74] in PD patients. Another study has demonstrated increased ICF in migrainers[75] indicating hyper responsivity of the glutamatergic intracortical circuits in these patients. In patients with depression, studies have shown that ICF is increased.[15,16] Further, another study[57] in children and adolescents with depression significant negative correlations were observed between Children’s Revised Depression Rating Scale and Quick Inventory of Depressive Symptomatology with bilateral ICF suggesting the impairment of glutamatergic circuits in early life depression. Impaired ICF was also observed in patients with non-AD mild cognitive impairment (MCI) while comparing to those with AD-MCI[76] indicating impairment in glutamatergic circuits in patients with non-AD MCI. However, ICF found to be normal in patients with AD,[77-79] the reproducibility of this measure (ICF) was lowest among other in patients with various disorders including AD.[80] In epilepsy, a meta-analysis showed increased ICF which is normalised with cathodal tDCS[81] and these intracortical circuit measure could be utilised as a marker of seizure control and duration of this disorder.[82] In patients with chronic cocaine usage, higher ICF has been observed than controls and task-evoked BOLD signal in the motor cortex was significantly correlated with ICF in the cocaine users suggesting potentially fruitful and treatable target in addiction.[19] There is selective increase in ICF after ethanol withdrawal.[62]
There are other intracortical circuits which could be measures using TMS such as short latency ICF (SICF), interhemispheric inhibition, inhibition of the motor cortex by cerebellar stimulation (cerebellar inhibition), short latency afferent inhibition (LAI) and long LAI. Further, TMS can also be used to explore interaction between two cortical regions such as dorsal premoror and primary motor cortex (dPM-M1), posterior parietal cortex and M1, primary sensory and motor cortex (SI-M1) and many more. Discussions about these circuits would be out of scope of this review and possibly covered in future communications, if required. Some of these intracortical circuits could be used in the evaluation of neurological and psychiatric disorders as summarised in [Table 2] which depicts the properties of different forms of cortical inhibition and facilitation [Figure 3] and their finding in several common movement and mood disorders.
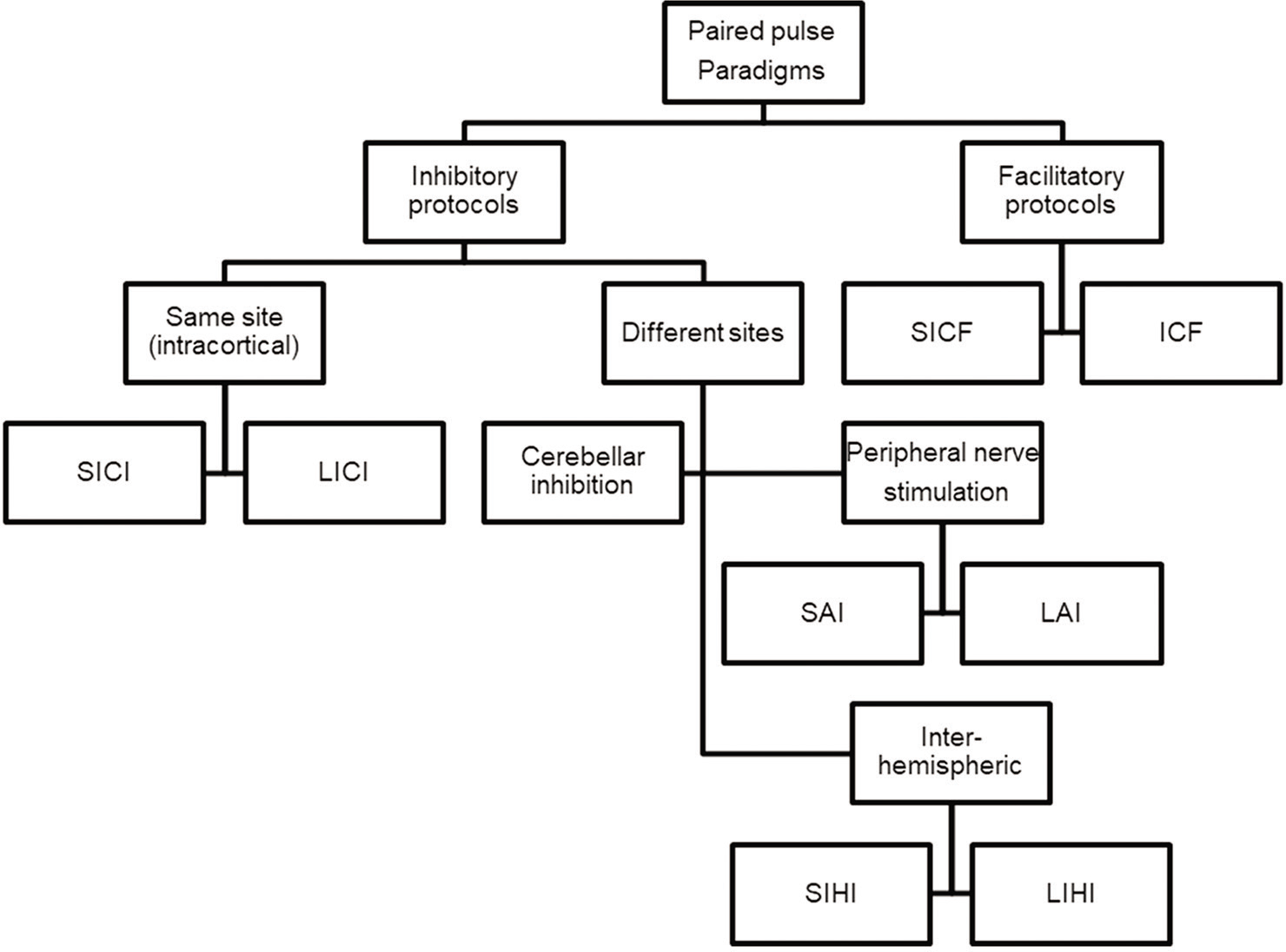
- Classification of different paired pulse paradigms (adapted from Ref 90) CBI: Cerebellar inhibition, ICF: Intracortical facilitation, LICI: Long interval intracortical inhibition, LIHI: Long interval interhemispheric inhibition, LAI: Long latency afferent inhibition, SAI: Short latency afferent inhibition, SICI: Short interval intracortical inhibition, SICF: Short interval intracortical facilitation, SIHI: Short interval interhemispheric inhibition.
PLASTICITY-LONG-TERM POTENTIATION (LTP) AND LONG-TERM DEPRESSION (LTD)
Neuronal plasticity refers to the ability of the neuron to modify its structure or functions in response to stimuli and these modifications outlast the stimulation period.[2] These changes generally occur in the synaptic functions, thus modifying the interneuron connections and are termed synaptic plasticity.[83,84] These changes encompass all possible mechanisms of neuronal network reorganisation, including recruitment of pathways that are functionally homologous but anatomically distinct from the original ones, reinforcement of existing synaptic connections, dendritic arborisation and synaptogenesis.[85] Such stimulation-induced modifications in synaptic efficacy, such as LTP and LTD, represent key cellular substrates for adaptive motor control and procedural memory as demonstrated in animal models.[86-88] LTP is, generally, defined as long-lasting but not necessarily irreversible increase in synaptic strength and LTD refers to decrease in synaptic strength. Induction of LTP and LTD depends on N-methyl D-Aspartate receptor activation by glutamate and post-synaptic calcium influx. A rapid increase in postsynaptic calcium concentration binds the C-terminal of calmodulin and triggers a kinase pathway that increases the density and conductance surface α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors leading to LTP. In contrast, a slower increase in calcium concentration promotes binding to the N-terminal of calmodulin, which operates through the phosphatase pathway and has opposite effect on surface AMPA receptors leading to LTD.[83,84,89]
MEASUREMENT OF MOTOR CORTICAL PLASTICITY USING TMS
Non-invasive brain stimulation technique such as TMS has been used to quantify various neurophysiologic measures in neurological and psychiatric disorders and has the potential to be used as a diagnostic and prognostic tool.[90] Various TMS protocols have been used to induce LTP- and LTD like changes in the brain and they may have therapeutic utilities in movement and mood disorders. Depending on the direction of change in excitability, these protocols have been broadly divided into LTP-like and LTD-like protocols, which increase or decrease the excitability. Protocols such as intermittent theta burst stimulation (iTBS), high frequency rTMS and paired associative stimulation (PAS) at 25 ms (N20 latency+~3ms; PAS25) are considered LTP-like protocols.[2,91] On the other hand, continuous (c) TBS, low frequency rTMS and PAS10 (PAS with 10 ms between peripheral nerve stimulation and M1-TMS) are considered LTD-like protocols as they decrease the excitability of the motor cortex.[2,91] However, the mechanisms by which these protocols induce the specific type of plasticity (LTP or LTD) are different. Although PAS25, high frequency rTMS and iTBS induced LTP-like changes in M1, the process of induction of plasticity, time courses and the mechanisms involved are different. In general, plasticity induced by protocols that activate multiple sets of synapses (such as PAS acting through sensory-motor communications and intracortical circuits of M1) is termed as heterosynaptic plasticity.[92] This type of plasticity depends on spike-timing dependent mechanisms of activating pre- and post-synaptic terminals within a time window as discussed earlier. This is different from homosynaptic plasticity (e.g., rTMS and TBS), which is induced by stimulating the same set of synaptic connections repeatedly and the effects are related to the frequency of stimulation. Although the molecular mechanisms of these non-invasive brain stimulation protocols involving homo and heterosynaptic plasticity[93] have not been elucidated, we could infer their mechanisms based on similar protocols in slice preparations in animal models. Furthermore, the effects of sensory inputs are altered in PD. Since sensory input in terms of peripheral stimulation is required for PAS and not so for rTMS, M1 plasticity probed by PAS and rTMS protocols may have different results.
PAIRED ASSOCIATIVE STIMULATION PROTOCOL
This involves repeated pairing (90–270 times) of peripheral nerve (say median nerve at the wrist) and M1-TMS with an ISI of around 25 ms ([time required for the sensory stimulus to reach sensory cortex is around 20ms-N20] and 3–5 ms for sensorimotor interaction) every 5–10 s.[2] This plasticity protocol induces LTP-like changes in M1 lasting for few minutes to hours. When PAS was used to assess the M1 plasticity in PD patients, impairment of plasticity was observed in most studies. However, studies have shown exaggerated plasticity on the less affected side and this exaggerated plasticity was associated with less severe clinical involvement. Thus, this increased plasticity may represent compensatory changes on the less affected side in early PD.[83] Exaggerated plasticity was also seen in tremulous patients with ‘scans without evidence of dopaminergic deficit (SWEDD), ’ compared to deficient plasticity observed in PD patients with dopaminergic deficit.[94] This study suggests that SWEDD patients may be closer to dystonia in terms of electrophysiological response to plasticity protocols than PD. Hence, plasticity response may differ in PD depending on subgroups and stages of PD.[83]
Dopaminergic medications modulate the altered plasticity in PD.[95] This restoration correlated with decreased plasticity and disease severity as measured by UPDRS scores. Furthermore, only PD patients without dyskinesias showed restoration of M1-plasticity by L-dopa.[96] Kishore et al. showed that the deficient PAS-induced plasticity in PD patients with LID is restored by inhibitory cTBS to the cerebellum.[97] The authors showed this restoration may be due to modulation of sensory input as only PAS but not iTBS induced M1 plasticity impairment was restored by cerebellar cTBS. In patients with LID treated with STN DBS, optimal stimulation in the on medication condition also restored PAS induced M1 plasticity to normal level.[98] Thus, M1 plasticity in PD changes with the phase of the disease with compensatory exaggeration on the unaffected side in early PD to deficient plasticity in later stages of PD, which may be restored with dopaminergic drugs and STN DBS. Further studies are required to elucidate the mechanisms and circuits involved in these issues of plasticity in PD. The results of M1 plasticity in PD using other protocols (TBS and rTMS) protocols are variable.[83]
THERAPEUTIC STRATEGIES THAT MODULATE M1 PLASTICITY BY NON-INVASIVE BRAIN STIMULATION
A study showed decreased activity in the mesial motor areas such as the supplementary motor area (SMA) in early stages of PD whereas hyperactivity was found in more lateral regions such as the M1 in more advanced stages of the disease.[99] Non-invasive brain stimulation techniques that alter the plasticity of these cortical-subcortical networks have been tested as treatment of PD. In early PD, rTMS in single and multiple session designs as well as anodal tDCS showed variable improvement in PD symptoms.[83] Thus, modulation of M1 excitability in PD has therapeutic potential. This may be further explored with pairing M1 stimulation with stimulation of basal ganglia structures such as STN and GPi DBS, other cortical structures (SMA, DLPFC and other cortical areas involved in PD) to further increase the clinical benefits. Furthermore, future studies that investigate other neurotransmitter pathways such as cholinergic, adrenergic and serotonergic systems with pharmacological agents and neuroimaging techniques will further our understanding of the pathophysiology of BG and M1 synaptic plasticity in PD and other neuropsychiatric disorders. This will help to develop new modes of investigations to further understand the disease and identify therapeutic targets for effective management of PD.
Dystonia and plasticity
In dystonia, plasticity studies have demonstrated exaggerated plasticity[67,91] which is responsible for task-specific dystonia such as writer’s or musician’s cramp as well as generalised or segmental dystonia. In all these conditions, co-contraction of both agonist and antagonist group of muscles is the common phenomenon which could be either due to decreased inhibition of motor cortical circuits[12,46] or exaggerated plasticity[67,91] in the M1-corticospinal tract.
Depression and plasticity
rTMS therapy has been approved by FDA to the treatment of major depression.[100] Although many studies explored the efficacy of different protocols of rTMS in depression, the clear understanding of factors which predict the improvement is not clearly defined. Some of the measures such as imaging, clinical phenotype, cardiac autonomic functions, balance of biochemical measures of inhibitory and excitatory neurotransmitters have been implicated as possible neurobiological measures predicting the efficacy of rTMS in treatment resistant depression. One of such studies showed higher functional connectivity between the left DLPFC and striatum predicted better treatment response to rTMS in patients with treatment resistant depression.[101] A study found elevated cortical plasticity which was associated with diminished cognitive inhibition in patients with late life depression suggesting that hyper-excitability of cortical circuits following repeated cortical activation which, in turn, may promote inappropriate pre-potent responses. This altered cortical plasticity might represent a neural mechanism underlying an inhibitory control cognitive endophenotype in this clinical condition.[102]
Schizophrenia and plasticity
Similar to the earlier explained concept of differential hemispheric threshold and other intracortical circuits,[17,18,42] asymmetric impairment of cortical plasticity the left parietal-frontal network in SCZ patients explored using PAS protocols.[103,104] Further studies need to explore the therapeutic utility of this phenomenology using NIBS such as tDCS for auditory hallucinations.[105]
Chronic pain and fibromyalgia
There have been many studies exploring utility of rTMS in chronic pain disorders including fibromyalgia.[106-108] Analgesic effects of rTMS were first reported in late 90’s using high frequency stimulation on precentral cortical regions.[109] Since then many conditions of chronic pain such as neuropathic pain, fibromyalgia and regional pain syndromes have been explored using various protocols and stimulation regions.[110] In addition to the pain many issues such as fatigue, depression, cognitive capabilities and quality of life has been improve with rTMS therapy. In addition to plasticity effects, activating neurons, increasing the blood supply and metabolism in the target regions, placebo effects of rTMS could be the mechanisms of action in these disorders. However, more systematic studies using sham stimulation protocols, different stimulation protocols and target regions need to be carried out in various chromic pain conditions.
LIMITATIONS OF TMS, SIDE-EFFECTS AND COMPARISON WITH OTHER NON-INVASIVE TECHNIQUES
Major limitations of TMS include higher cost of the equipment set-up (few lakhs to crores of Rupees), hands-on training required for delivery and analysis and not many large public studies done in India. All these issues are interdependent as because of its high cost related issues not many centres are housing this equipment leading to few trained hands in the country. This, in turn, leads to not many single or multicentric studies coming out from our country. Non-availability of Indian made equipment (which is currently being manufactured at Europe) is the major reason for the high cost. Our electrical engineers need to look into the basic design of the apparatus and possibly prepare indigenous equipment under ‘Make in India’ flagship program. We also need to develop collaboration between the institutions, disciplines and scientists to explore further the utility of TMS in exploring neuroscientific field.
Some side effects of TMS were reported such as light headache (mainly due to scalp muscle contraction during stimulation which usually fades off after stimulation or by rest or pain medications), tinnitus or transient hearing loss (due to clicking/tapping sounds which could be avoided by ear plugs) and rare reports of epileptic seizures during high frequency stimulation. Most of these could be avoided by preselecting subjects/patients (avoiding subjects with history of epilepsy, metallic implants or pacemakers in head and neck area) before the study and thorough history and physical examination and providing proper care during and after stimulation.
In addition to TMS, other NIBS techniques such as tDCS, transcranial alternating current stimulation (tACS), random noise stimulation, transcutaneous auricular vagal nerve stimulation and ECT are currently in use. Discussions as to compare these techniques with TMS and listing advantages and disadvantages of each technique are beyond the scope of this review and probably will be covered in a future review on NIBS techniques. Although, these other NIBS techniques cheaper than TMS, these will not help in assessing the motor excitability as MEP cannot be triggered with any of these techniques except tACS (which is very painful on stimulation). However, combining different modes of NIBS to achieve the optimal benefits is currently being undertaken.[111-113]
FUTURE DIRECTIONS
TMS and other non-invasive techniques have enabled to advance the understanding the complex circuitry of human brain. These non-invasive investigative tools (alone or in combination) coupled with advanced imaging techniques and other measures of neuroinvestigation would throw more light into the neurophysiological mechanisms of neurological and psychiatric disorders and to design potential therapeutic options. This field of NIBS is still naïve in India and we need more studies and centres coming up with many indigenously developed stimulation equipment.
CONCLUSION
TMS is a non-invasive, painless technique to stimulate the human brain. Studies have demonstrated utility of TMS in understanding the cortical neuronal circuits as well as treating few of the neurological and psychiatric disorders. However, there is necessity of more clinical research studies especially from India to explore further investigative and therapeutic implications of TMS. Hopefully, this review would elicit some interest among physiologists to take up this exciting area of brain stimulation and work further in collaboration with clinicians exploring efficacy of TMS as investigative and treatment modality for various brain-related disorders.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Non-invasive magnetic stimulation of human motor cortex. Lancet. 1985;1:1106-7.
- [CrossRef] [Google Scholar]
- Measurement and modulation of plasticity of the motor system in humans using transcranial magnetic stimulation. Motor Control. 2009;13:442-53.
- [CrossRef] [PubMed] [Google Scholar]
- Transcranial magnetic stimulation: A primer. Neuron. 2007;55:187-99.
- [CrossRef] [PubMed] [Google Scholar]
- The clinical diagnostic utility of transcranial magnetic stimulation: Report of an IFCN committee. Clin Neurophysiol. 2007;119:504-32.
- [CrossRef] [PubMed] [Google Scholar]
- Applications of transcranial magnetic stimulation in movement disorders. J Clin Neurophysiol. 2002;19:272-93.
- [CrossRef] [PubMed] [Google Scholar]
- Studies of human motor physiology with transcranial magnetic stimulation. Muscle Nerve Suppl. 2000;9:S26-32.
- [CrossRef] [Google Scholar]
- Cortico-motor excitability of the lower limb motor representation: A comparative study in Parkinson's disease and healthy controls. Clin Neurophysiol. 2002;113:2006-12.
- [CrossRef] [Google Scholar]
- Abnormal facilitation of the response to transcranial magnetic stimulation in patients with Parkinson's disease. Neurology. 1994;44:735-41.
- [CrossRef] [PubMed] [Google Scholar]
- Usefulness of transcranial magnetic stimulation to assess motor function in patients with parkinsonism. Ann Rehabil Med. 2016;40:81-7.
- [CrossRef] [PubMed] [Google Scholar]
- Alterations in cortical excitability and central motor conduction time in spinocerebellar ataxias 1, 2 and 3: A comparative study. Parkinsonism Relat Disord. 2013;19:306-11.
- [CrossRef] [PubMed] [Google Scholar]
- Hypo-excitability of cortical areas in patients affected by Friedreich ataxia: A TMS study. J Neurol Sci. 2005;235:19-22.
- [CrossRef] [PubMed] [Google Scholar]
- Elevated threshold for intracortical inhibition in focal hand dystonia. Mov Disord. 2004;19:1312-7.
- [CrossRef] [PubMed] [Google Scholar]
- Alterations of cortical excitability and central motor conduction time in Wilson's disease. Neurosci Lett. 2013;553:90-4.
- [CrossRef] [PubMed] [Google Scholar]
- The effect of daily prefrontal repetitive transcranial magnetic stimulation over several weeks on resting motor threshold. Brain Stimul. 2009;2:163-7.
- [CrossRef] [PubMed] [Google Scholar]
- Evidence for increased motor cortical facilitation and decreased inhibition in atypical depression. Acta Psychiatr Scand. 2016;134:172-82.
- [CrossRef] [PubMed] [Google Scholar]
- Modulation of motor cortex excitability predicts antidepressant response to prefrontal cortex repetitive transcranial magnetic stimulation In: Brain Stimul. Vol 10. 2017. p. :787-94.
- [CrossRef] [PubMed] [Google Scholar]
- Motor cortical excitability in schizophrenia. Biol Psychiatry. 2002;52:24-31.
- [CrossRef] [Google Scholar]
- A transcranial magnetic stimulation study of inhibitory deficits in the motor cortex in patients with schizophrenia. Psychiatry Res. 2002;114:11-22.
- [CrossRef] [Google Scholar]
- A comprehensive study of sensorimotor cortex excitability in chronic cocaine users: Integrating TMS and functional MRI data. Drug Alcohol Depend. 2015;157:28-35.
- [CrossRef] [PubMed] [Google Scholar]
- Central motor conduction time. Handb Clin Neurol. 2013;116:375-86.
- [CrossRef] [PubMed] [Google Scholar]
- Increased cortical excitability with longer duration of Parkinson's disease as evaluated by transcranial magnetic stimulation. Neurol India. 2003;51:13-5.
- [Google Scholar]
- Sensory and motor evoked potentials in multiple system atrophy: A comparative study with Parkinson's disease. Mov Disord. 1997;12:315-21.
- [CrossRef] [PubMed] [Google Scholar]
- The corticomotoneurone connection is normal in Parkinson's disease. Nature. 1984;310:407-9.
- [CrossRef] [PubMed] [Google Scholar]
- Parkinson's disease rigidity: Magnetic motor evoked potentials in a small hand muscle. Neurology. 1991;91:1449-56.
- [CrossRef] [PubMed] [Google Scholar]
- Central motor conduction time in Parkinson's disease. J Clin Neurosci. 1999;6:17-9.
- [CrossRef] [Google Scholar]
- Abnormalities of central motor conduction in Parkinson's disease. J Neurol Sci. 1990;100:94-7.
- [CrossRef] [Google Scholar]
- Effect of therapy on motor cortical excitability in Parkinson's disease. Can J Neurol Sci. 2008;35:166-72.
- [CrossRef] [PubMed] [Google Scholar]
- Motor cortical physiology in patients and asymptomatic carriers of parkin gene mutations. Mov Disord 208;. ;23:1812-9.
- [CrossRef] [PubMed] [Google Scholar]
- Neurophysiological evaluation of motor corticospinal pathways by TMS in idiopathic early-onset Parkinson's disease. Clin Neurophysiol. 2011;122:546-9.
- [CrossRef] [PubMed] [Google Scholar]
- Cognitive impairment and central motor conduction time in chronic alcoholics. Funct Neurol. 2002;17:83-6.
- [Google Scholar]
- Continuous intrathecal baclofen infusions induced a marked increase of the transcranially evoked silent period in a patient with generalized dystonia. Muscle Nerve. 1998;21:1209-12.
- [CrossRef] [Google Scholar]
- Differential effects on motorcortical inhibition induced by blockade of GABA uptake in humans. J Physiol. 1999;517:591-7.
- [CrossRef] [PubMed] [Google Scholar]
- Shortened silent period produced by magnetic cortical stimulation in patients with Parkinson's disease. J Neurol Sci. 1995;130:209-14.
- [CrossRef] [Google Scholar]
- Motor cortical inhibition and the dopaminergic system. Pharmacological changes in the silent period after transcranial brain stimulation in normal subjects. patients with Parkinson's disease and drug-induced parkinsonism. Brain. 1994;117:317-23.
- [CrossRef] [PubMed] [Google Scholar]
- Effects of internal globus pallidus stimulation on motor cortex excitability. Neurology. 2001;56:716-23.
- [CrossRef] [PubMed] [Google Scholar]
- The relationship between cortical inhibition and electroconvulsive therapy in the treatment of major depressive disorder. Sci Rep. 2016;6:37461.
- [CrossRef] [PubMed] [Google Scholar]
- Increased cortical inhibition in depression: A prolonged silent period with transcranial magnetic stimulation (TMS) Psychol Med. 2000;30:565-70.
- [CrossRef] [PubMed] [Google Scholar]
- Cortical inhibition in major depression: Investigating the acute effect of single-session yoga versus walking. Brain Stimul. 2019;12:1597-9.
- [CrossRef] [PubMed] [Google Scholar]
- Deficient inhibitory cortical networks in antipsychotic-naive subjects at risk of developing first-episode psychosis and first-episode schizophrenia patients: A cross-sectional study. Biol Psychiatry. 2012;72:744-51.
- [CrossRef] [PubMed] [Google Scholar]
- The relationship between motor cortex excitability and severity of Alzheimer's disease: A transcranial magnetic stimulation study. Clin Neurophysiol. 2011;41:107-13.
- [CrossRef] [PubMed] [Google Scholar]
- Modulation of human motor cortex excitability by quetiapine. Psychopharmacology (Berl). 2008;196:623-9.
- [CrossRef] [PubMed] [Google Scholar]
- Motor circuit abnormalities in first-episode schizophrenia assessed with transcranial magnetic stimulation. Pharmacopsychiatry. 2009;42:194-201.
- [CrossRef] [PubMed] [Google Scholar]
- Effects of motor imagery on motor cortical output topography in Parkinson's disease. Neurology. 2001;57:55-61.
- [CrossRef] [PubMed] [Google Scholar]
- The effect of transcranial magnetic stimulation on median nerve somatosensory evoked potentials. Electroencephalogr Clin Neurophysiol. 1993;89:227-34.
- [CrossRef] [Google Scholar]
- Changes in excitability of motor cortical circuitry in patients with Parkinson's disease. Ann Neurol. 1995;37:181-8.
- [CrossRef] [PubMed] [Google Scholar]
- Short intracortical and surround inhibition are selectively reduced during movement initiation in focal hand dystonia. J Neurosci. 2008;28:10363-9.
- [CrossRef] [PubMed] [Google Scholar]
- Cortical excitability in very mild Alzheimer's disease: A long-term follow-up study. J Neurol. 2010;257:2078-85.
- [CrossRef] [PubMed] [Google Scholar]
- Association of cortical disinhibition with tic, ADHD, and OCD severity in Tourette syndrome. Mov Disord. 2004;19:416-25.
- [CrossRef] [PubMed] [Google Scholar]
- Increased motor cortical facilitation and decreased inhibition in Parkinson disease. Neurology. 2013;80:1746-53.
- [CrossRef] [PubMed] [Google Scholar]
- Pathways mediating abnormal intracortical inhibition in Parkinson's disease. Ann Neurol. 2005;58:516-24.
- [CrossRef] [PubMed] [Google Scholar]
- Facilitatory/inhibitory intracortical imbalance in REM sleep behavior disorder: Early electrophysiological marker of neurodegeneration? Sleep. 2020;43:zsz242.
- [CrossRef] [PubMed] [Google Scholar]
- Reversal of cortical reorganization in human primary motor cortex following thumb reconstruction. J Neurophysiol. 2010;103:65-73.
- [CrossRef] [PubMed] [Google Scholar]
- Antidepressant-resistant depression is characterized by reduced short-and long-interval cortical inhibition. Psychol Med. 2020;50:1285-91.
- [CrossRef] [PubMed] [Google Scholar]
- Transcranial magnetic stimulation markers of antidepressant treatment in adolescents with major depressive disorder. Int J Neuropsychopharmacol. 2019;22:435-44.
- [CrossRef] [PubMed] [Google Scholar]
- A meta-analysis of cortical inhibition and excitability using transcranial magnetic stimulation in psychiatric disorders. Clin Neurophysiol. 2013;124:1309-20.
- [CrossRef] [PubMed] [Google Scholar]
- Preliminary evidence of an association between increased cortical inhibition and reduced suicidal ideation in adolescents treated for major depression. J Affect Disord. 2019;244:21-4.
- [CrossRef] [PubMed] [Google Scholar]
- Cortical inhibitory and excitatory correlates of depression severity in children and adolescents. J Affect Disord. 2016;190:566-75.
- [CrossRef] [PubMed] [Google Scholar]
- Association of intracortical inhibition with social cognition deficits in schizophrenia: Findings from a transcranial magnetic stimulation study. Schizophr Res. 2014;158:146-50.
- [CrossRef] [PubMed] [Google Scholar]
- Reduction of cortical GABAergic inhibition correlates with working memory impairment in recent onset schizophrenia. Schizophr Res. 2013;146:238-43.
- [CrossRef] [PubMed] [Google Scholar]
- Changes in motor cortical excitability in schizophrenia following transcranial direct current stimulation. Prog Neuropsychopharmacol Biol Psychiatry. 2019;90:43-8.
- [CrossRef] [PubMed] [Google Scholar]
- Differential effects of cannabis dependence on cortical inhibition in patients with schizophrenia and non-psychiatric controls. Brain Stimul. 2017;10:275-82.
- [CrossRef] [PubMed] [Google Scholar]
- Altered motor cortex excitability to magnetic stimulation in alcohol withdrawal syndrome. Alcohol Clin Exp Res. 2010;34:628-32.
- [CrossRef] [PubMed] [Google Scholar]
- GABA-ergic tone hypothesis in hepatic encephalopathy-revisited. Clin Neurophysiol. 2019;130:911-6.
- [CrossRef] [PubMed] [Google Scholar]
- Segregating two inhibitory circuits in human motor cortex at the level of GABAA receptor subtypes: A TMS study. Clin Neurophysiol. 2007;118:2207-14.
- [CrossRef] [PubMed] [Google Scholar]
- Cortical inhibition in Parkinson's disease. A study with paired magnetic stimulation. Brain. 1996;119:71-7.
- [CrossRef] [PubMed] [Google Scholar]
- Motor evoked responses to paired cortical magnetic stimulation in Parkinson's disease. Electroencephalogr Clin Neurophysiol. 1997;105:37-43.
- [CrossRef] [Google Scholar]
- Motor cortical circuits in Parkinson disease and dystonia. Handb Clin Neurol. 2019;161:167-86.
- [CrossRef] [PubMed] [Google Scholar]
- Effect of apomorphine on cortical inhibition in Parkinson's disease patients: A transcranial magnetic stimulation study. Exp Brain Res. 2001;141:52-62.
- [CrossRef] [PubMed] [Google Scholar]
- Interactions between two different inhibitory systems in the human motor cortex. J Physiol. 2001;530:307-17.
- [CrossRef] [PubMed] [Google Scholar]
- Cortical inhibition in symptomatic and remitted mania compared to healthy subjects: A cross-sectional study. Bipolar Disord. 2017;19:698-703.
- [CrossRef] [PubMed] [Google Scholar]
- Differential pharmacological effects on brain reactivity and plasticity in Alzheimer's disease. Front Psychiatry. 2013;4:124.
- [CrossRef] [PubMed] [Google Scholar]
- Investigating cortical inhibition in first-degree relatives and probands in schizophrenia. Sci Rep. 2017;7:43629.
- [CrossRef] [PubMed] [Google Scholar]
- Intracortical inhibition and facilitation are impaired in patients with early Parkinson's disease: A paired TMS study. Eur J Neurol. 2003;10:385-9.
- [CrossRef] [PubMed] [Google Scholar]
- Transcranial magnetic stimulation follow-up study in early Parkinson's disease: A decline in compensation with disease progression? Mov Disord. 2015;30:1098-106.
- [CrossRef] [PubMed] [Google Scholar]
- Intracortical facilitation within the migraine motor cortex depends on the stimulation intensity. A paired-pulse TMS study. J Headache Pain. 2018;19:65.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis of mild cognitive impairment due to Alzheimer's disease with transcranial magnetic stimulation. J Alzheimers Dis. 2018;65:221-30.
- [CrossRef] [PubMed] [Google Scholar]
- Intra-and inter-cortical motor excitability in Alzheimer's disease. J Neural Transm (Vienna). 2012;119:605-12.
- [CrossRef] [PubMed] [Google Scholar]
- Different TMS patterns of intracortical inhibition in early onset Alzheimer dementia and frontotemporal dementia. Clin Neurophysiol. 2004;115:2410-8.
- [CrossRef] [PubMed] [Google Scholar]
- The contribution of TMS to frontotemporal dementia variants. Acta Neurol Scand. 2008;118:275-80.
- [CrossRef] [PubMed] [Google Scholar]
- Reproducibility of single-pulse, paired-pulse, and intermittent theta-burst TMS measures in healthy aging, Type-2 diabetes, and Alzheimer's disease. Front Aging Neurosci. 2017;9:263.
- [CrossRef] [PubMed] [Google Scholar]
- The effects of transcranial direct current stimulation on short-interval intracortical inhibition and intracortical facilitation: A systematic review and meta-analysis. Rev Neurosci. 2018;29:99-114.
- [CrossRef] [PubMed] [Google Scholar]
- Cortical excitability correlates with seizure control and epilepsy duration in chronic epilepsy. Ann Clin Transl Neurol. 2017;4:87-97.
- [CrossRef] [PubMed] [Google Scholar]
- Motor cortical plasticity in Parkinson's disease. Front Neurol. 2013;4:128.
- [CrossRef] [PubMed] [Google Scholar]
- Synaptic plasticity of NMDA receptors: Mechanisms and functional implications. Curr Opin Neurobiol. 2012;22:496-508.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular mechanisms that underlie structural and functional changes at the postsynaptic membrane during synaptic plasticity. Prog Neurobiol. 1998;55:611-40.
- [CrossRef] [Google Scholar]
- AMPA receptor regulation during synaptic plasticity in hippocampus and neocortex. Semin Cell Dev Biol. 2011;22:514-20.
- [CrossRef] [PubMed] [Google Scholar]
- Discovering long-term potentiation (LTP)-recollections and reflections on what came after. Acta Physiol (Oxf). 2018;222:1.
- [CrossRef] [PubMed] [Google Scholar]
- The discovery of long-term potentiation. Philos Trans R Soc Lond B Biol Sci. 2003;358:617-20.
- [CrossRef] [PubMed] [Google Scholar]
- Basic roles of key molecules connected with NMDAR signaling pathway on regulating learning and memory and synaptic plasticity. Mil Med Res. 2016;3:26.
- [CrossRef] [PubMed] [Google Scholar]
- Single pulse TMS, Paired pulse TMS and rTMS In: Kompoliti K, Metman LV, eds. Encyclopedia of Movement Disorders. Vol 3. Oxford: Academic Press; 2010. p. :116-8, 354-8, 53-5
- [CrossRef] [Google Scholar]
- Task-specific hand dystonia: Can too much plasticity be bad for you? Trends Neurosci. 2006;29:192-9.
- [CrossRef] [PubMed] [Google Scholar]
- Heterosynaptic plasticity: Multiple mechanisms and multiple roles. Neuroscientist. 2014;20:483-98.
- [CrossRef] [PubMed] [Google Scholar]
- Mechanisms of heterosynaptic metaplasticity. Philos Trans R Soc Lond B Biol Sci. 2013;369:20130148.
- [CrossRef] [PubMed] [Google Scholar]
- Distinguishing SWEDDs patients with asymmetric resting tremor from Parkinson's disease: A clinical and electrophysiological study. Mov Disord. 2010;25:560-9.
- [CrossRef] [PubMed] [Google Scholar]
- Motor cortex plasticity in Parkinson's disease and levodopa-induced dyskinesias. Brain. 2006;129:1059-69.
- [CrossRef] [PubMed] [Google Scholar]
- Cortical plasticity and levodopa-induced dyskinesias in Parkinson's disease: Connecting the dots in a multicomponent network. Clin Neurophysiol. 2017;128:992-9.
- [CrossRef] [PubMed] [Google Scholar]
- Cerebellar sensory processing alterations impact motor cortical plasticity in Parkinson's disease: Clues from dyskinetic patients. Cereb Cortex. 2014;24:2055-67.
- [CrossRef] [PubMed] [Google Scholar]
- Effects of subthalamic nucleus stimulation on motor cortex plasticity in Parkinson disease. Neurology. 2015;85:425-32.
- [CrossRef] [PubMed] [Google Scholar]
- Theta burst stimulation over the supplementary motor area in Parkinson's disease. J Neurol. 2015;262:357-64.
- [CrossRef] [PubMed] [Google Scholar]
- The clinical TMS society consensus review and treatment recommendations for TMS therapy for major depressive disorder. Brain Stimul. 2016;9:336-46.
- [CrossRef] [PubMed] [Google Scholar]
- Functional connectivity of the left DLPFC to striatum predicts treatment response of depression to TMS. Brain Stimul. 2017;10:919-25.
- [CrossRef] [PubMed] [Google Scholar]
- An inverse relationship between cortical plasticity and cognitive inhibition in late-life depression. Neuropsychopharmacology. 2019;44:1659-66.
- [CrossRef] [PubMed] [Google Scholar]
- Left hemispheric breakdown of LTP-like cortico-cortical plasticity in schizophrenic patients. Clin Neurophysiol. 2017;128:2037-42.
- [CrossRef] [PubMed] [Google Scholar]
- Transcranial direct current stimulation in schizophrenia. Clin Psychopharmacol Neurosci. 2013;11:118-25.
- [CrossRef] [PubMed] [Google Scholar]
- Efficacy of transcranial direct current stimulation and repetitive transcranial magnetic stimulation for treating fibromyalgia syndrome: A systematic review. Pain Pract. 2013;13:131-45.
- [CrossRef] [PubMed] [Google Scholar]
- Non-invasive brain stimulation techniques for chronic pain. Cochrane Database Syst Rev. 2014;4:CD008208.
- [CrossRef] [PubMed] [Google Scholar]
- Repetitive transcranial magnetic stimulation in chronic pain: A review of the literature. Arch Phys Med Rehabil. 2015;96:S156-72.
- [CrossRef] [PubMed] [Google Scholar]
- Chronic pain treated by rTMS of motor cortex. Electroencephalogr Clin Neurophysiol. 1998;107:92.
- [Google Scholar]
- Is rTMS a therapeutic option in chronic pain syndrome? Insights from the treatment of fibromyalgia. Pain. 2011;152:1447-8.
- [CrossRef] [PubMed] [Google Scholar]
- Dual stimulation with tDCS-iTBS as add-on treatment in recurrent depressive disorder-a case report. Brain Stimul. 2020;13:625-6.
- [CrossRef] [PubMed] [Google Scholar]
- Modulation of iTBS after-effects via concurrent directional TDCS: A proof of principle study. Brain Stimul. 2017;10:744-7.
- [CrossRef] [PubMed] [Google Scholar]
- Direct-current-dependent shift of theta-burst-induced plasticity in the human motor cortex. Exp Brain Res. 2012;217:15-23.
- [CrossRef] [PubMed] [Google Scholar]




