
Translate this page into:
Molecular mechanisms of α7-nAchR-mediated anti-inflammatory effects
*Corresponding author: Mahmoud Elsaid Youssef, Department of Pharmacology and Biochemistry, Delta University for Science and Technology, International Coastal road, Mansoura 35511, Dakahlia, Egypt. mahmoodelsaid@hotmail.com
-
Received: ,
Accepted: ,
How to cite this article: Youssef ME, Moustafa Y, Abdelrazek H. Molecular mechanisms of α7-nAchR-mediated anti-inflammatory effects. Indian J Physiol Pharmacol 2020;64(3):158-73.
Abstract
The cholinergic anti-inflammatory pathway is described as an interaction between the nervous system and the immune system. This interaction is regulated by the α7 subtype of cholinergic nicotinic Ach receptors (α7-nAchR), which leads to a marked decrease in the inflammatory cytokines, such as interleukin (IL)-1β, IL-6 and tumour necrosis factor α. Several ligands that interact with α7-nAchR have been recently discovered. These ligands vary in their source, chemical structure, selectivity, potency and efficacy. Activation of α7-nAchR either selectively or non-selectively showed an anti-inflammatory effect that could be due to the inhibition of inflammatory signalling pathways such as Toll-like receptor 4/nuclear factor kappa B inflammasome and mammalian target of rapamycin-mediated autophagy pathways. In addition, it was proved that continuous activation of α7-nAchR could stimulate several anti-inflammatory signalling mechanisms, including Janus activated kinase-2/signal transducer and activator of transcription 3, nuclear factor erythroid 2-related factor 2/HO-1 and adenosine monophosphate-activated protein kinase signalling. In this review, we focused on the recent discoveries of α7-nAchR agonists and antagonists and their anti-inflammatory mechanisms.
Keywords
α7-nAchR
Inflammation
Cholinergic anti-inflammatory pathway
Toll-like receptor 4
Nuclear factor kappa B
Janus activated kinase-2
Signal transducer and activator of transcription 3
Mammalian target of rapamycin
Nuclear factor erythroid 2-related factor 2
HO-1
Adenosine monophosphate-activated protein kinase

INTRODUCTION
The cholinergic anti-inflammatory pathway was subjected to many studies since its discovery. Catabolite activator protein (CAP) is described as an interaction that occurs among the nervous and the immune systems. CAP is believed to be involved in the regulation of controls the inflammatory process.[1] This pathway is initiated by the stimulation of the afferent vagus nerve either by lipopolysaccharides (LPS) or pro-inflammatory cytokines.[2] The vagal signal is processed centrally through a muscarinic-receptors dependent pathway.[3] The initiated anti-inflammatory signal reaches the celiac-superior mesenteric plexus through the efferent vagus nerve.[4] The anti-inflammatory signal is transmitted to the spleen with the aid of the splenic nerve that is connected to efferent vagus nerve fibres.[5] Norepinephrine is released at the terminal of the splenic nerve, leading to the activation of β2 receptors and an increase in the expression of choline acetyltransferase enzyme that synthesises acetylcholine (Ach) in specialised T-lymphocytes.[6] Activated T-lymphocytes release Ach in the spleen, which stimulates α7 nicotinic Ach receptors (α7-nAchR) in macrophages, which leads to a downregulation in the pro-inflammatory cytokines expression[7] without affecting the anti-inflammatory cytokines levels. CAP was previously studied in human monocytes after exposure to LPS. Treatment with nicotine led to a marked decrease in the plasma levels of inflammatory cytokines such as interleukin 1β (IL-1β), IL-6 and tumour necrosis factor α (TNF-α) in previously challenged human monocytes by LPS through activation of α7-nAchR,[8] whereas, the plasma levels of anti-inflammatory cytokines such as IL-10 were not affected.[9] In addition, the role of α7-nAchR was confirmed by subjecting monocytes with the knockout α7-nAchR gene to LPS.[10] These experiments have revealed the crucial role of α7-nAchR in regulating CAP. It is believed that the anti-inflammatory effect that mediated by α7-nAchR could be due the attenuation of several inflammatory pathways. This review article describes the different agonists and antagonists of α7-nAchR. In addition, we reviewed the recent studies of these drugs in different animal models, including neuronal and non-neuronal effects, which are based on the activation of α7-nAchR. Finally, this review illustrates the molecular mechanisms that mediate the anti-inflammatory effect of α7-nAchR. We have reviewed research articles that focus on studying the mechanisms by which α7-nAchR prevent inflammation in various animal models of inflammatory diseases.
α7-nAchR
nAchR are member of the Cys-loop ligand-gated ion channel superfamily, which also includes serotonin subtype 3 (5-HT3) receptors and γ-aminobutyric acid receptors (GABA).[11] nAchR participates in several physiological functions, as they control the contraction of skeletal muscles in the neuromuscular junction and regulate the ganglionic functions in the autonomic nervous system. In addition, nAchR are found in non-neuronal cells, such as endothelial cells, glial cells and immune cell where in these sites, α7-nAchR regulate anti-inflammatory and angiogenic responses. nAchR are formed by the assembly of five subunits that are arranged symmetrically in the form of a ring with a concentric channel through which selective cations pass.[12,13] Each subunit is composed of hydrophobic transmembrane domains specified as M1 through M4, an intracellular loop between M3 and M4 and extracellular N-terminal.[14] nAchR are widely distributed in the central nervous system, peripheral nervous system, muscles and many other tissues. They are the primary receptors in the neuromuscular junction for controlling skeletal muscle contraction. In the autonomic ganglia, they transmit signals from presynaptic neurons to post-synaptic neurons in both sympathetic and parasympathetic systems. Angiogenesis, in addition to the inflammatory response, is regulated by nAchR in blood vessels and immune cells by intracellular mechanisms.[15] nAchR subunits have been recognised and classified into muscle-type and neuronal-type subunits.[16] These subunits are subdivided into four subfamilies (I-IV) that are specified according to similarities in the protein sequence.[17] Besides, the subfamily III has been further divided into three subtypes [Table 1].
| Type | Neuronal-type | Muscle-type | ||||
|---|---|---|---|---|---|---|
| Subfamily | I | II | III | IV | ||
| i | ii | iii | ||||
| Subunit | α9 | α7 | α2 | β2 | β3 | α1 |
| α10 | α8 | α3 | β4 | α5 | β1 | |
| α4 | δ | |||||
| γ | ||||||
| ε | ||||||
The nAchR subunits are composed by the assembly of five subunits. This table shows the different 17 subunits, which comprise the different nAchR subfamilies
α7-nAchR are formed by the assemblage of five subunits of the α7 subtype. For this reason, α7-nAchR are considered homomeric receptors.[18] The α7-nAchR are expressed mainly in the hippocampus and prefrontal cortex, where they influence glutamatergic and GABAergic synapses.[19] There is substantial evidence for the contribution of α7-nAchR to synaptic potentiation of the hippocampus due to increased Ca++ permeability.[20] α7-nAchR participate in memory and learning. In addition, they have implications in some neurological disorders such as Parkinson’s disease, schizophrenia[21] and Alzheimer’s disease.[22] α7-nAchR are also expressed in immune cells such as lymphocytes, monocytes and macrophages their stimulation mediates CAP. In endothelial cells, furthermore, α7-nAchR are believed to regulate the angiogenesis process.
CLASSICAL AGONISTS
There is a high diversity of molecules that interact selectively and non-selectively with α7-nAchR [Table 2]. Therefore, a high number of α7-nAchR agonists and antagonists have been developed. α7-nAchR agonists are presented with structural diversity, whether they are separated naturally from animals or plants or they are completely synthesised. These well-known agonists are non-selective and differ in their potency and efficacy.[23] Ach is a non-selective endogenous agonist of all cholinergic receptors. However, its usage is confined due to the lack of selectivity between nicotinic and muscarinic receptors in addition to its short duration of action due to rapid hydrolysis. Atropine, a non-selective muscarinic antagonist, is usually given with Ach to improve its selectivity toward nAchR. However, choline, the metabolite of Ach hydrolysis, is a selective α7-nAchR agonist.[24] Anatoxin is a potent non-selective agonist that resembles Ach with 8 times more potency.[25] Carbachol, the carbamate analogue of Ach, is a weak nicotinic Ach agonist with more selectivity toward muscarinic receptors. It has a lower activity on α7-nAchR.[26]
| Drug | Selectivity | Activity | Key findings | Maximum in vivodose |
|---|---|---|---|---|
| Classical agonists and antagonists | ||||
| Ach | All muscarinic and nAchR | Agonist | ||
| Atropine | All muscarinic receptors | Antagonist | 0.04 mg/kg | |
| Choline | α7-nAchR | Agonist | 60 mg/kg | |
| Anatoxin | All nAchR | Agonist | 0.4 mg/kg | |
| Carbachol | nAchR Muscarinic receptors |
Weak agonist Strong agonist |
1 mg/kg | |
| Epibatidine | All nAchR | Strong agonist | ||
| Nicotine | All nAchR except α9-nAchR | Strong agonist | 400 µg/kg | |
| Cotinine | All nAchR | Weak agonist | ||
| Synthetic α7-nAchR agonists | ||||
| A-582941 | α7-nAchR | Partial agonist | • Improved negative symptoms of schizophrenia.[32] • Enhanced memory.[33] • Improved cognitive defects in schizophrenia.[31] |
10 mg/kg |
| A-844606 | α7-nAchR | Partial agonist | • Needs further investigations.[35] | |
| AR-R 17779 | α7-nAchR α4β2-nAchR α3β2-nAchR α3β4-nAchR |
Strong agonist Weak agonist Weak agonist Weak agonist |
• Cognitive enhancing activity[38,39] • Anti-inflammatory activity.[40] • Treatment of neurodegenerative diseases.[41] |
2 mg/kg |
| GTS-21 | α7-nAchR α4β2-nAchR |
Partial agonist Weak antagonist |
• Treatment of dementia.[44] • Improved attention in schizophrenia.[45] • Cognitive enhancement.[46] • Management of neurodegenerative diseases.[47] • Treating nicotine addiction.[50] • Improvement of Alzheimer’s disease.[51] • Anti-inflammatory.[53] • Attenuation of body wight loss.[56] • Reduces vascular permeability.[57] |
10 mg/kg |
| PHA-543613 | α7-nAchR | Strong agonist | • Neuroprotective activity.[59] • Treatment of Parkinson’s disease.[60] • Cognitive enhancement.[58,61] • Treatment of dementia.[62,63] • Angiogenic.[64] • Reducing neuropathic pain.[66] |
1 mg/kg |
| PNU-282987 | α7-nAchR 5-HT3 |
Strong agonist Weak agonist |
• Cognition enhancement.[68] • Treatment of Parkinson’s disease.[72] • Reduced dopaminergic neurons loss.[73] • Anti-inflammatory effect.[75] • Prevented prolonged febrile seizures.[74] • Prevented the damage of retinal ganglion.[82] |
30 mg/kg |
| Tropisetron | α7-nAchR α9α10 5-HT3 |
Partial agonist Strong antagonist Strong antagonist |
• Improved P50 auditory suppression in schizophrenia.[84] • Improved cognitive dysfunction.[86] • Ameliorated the disruption of dopaminergic neurons.[88] • Attenuated morphine withdrawal symptoms.[90] • Anti-inflammatory effect.[92] |
3 mg/kg |
| Antagonists of α7-nAchR | ||||
| α-bungarotoxin | α1β1γεδ-nAchR α7-nAchR α9 nAchR |
Strong antagonist Strong antagonist Strong antagonist |
||
| Methyllycaconitine | α7-nAchR α9-nAchR α9α10-nAchR α1β1γεδ-nAchR |
Strong antagonist Strong antagonist Strong antagonist Weak antagonist |
10 mg/kg | |
Another well-established, very potent, non-selective agonist is epibatidine. It showed α7-nAchR activity in the micromolar range.[27] Nicotine is the most common non-selective nAchR agonist. It activates all nAchR, except α9 receptors, at micromolar concentrations.[28] Nicotine is an alkaloid separated from tobacco and has been historically used to differentiate between different nAchR. The lipid solubility of nicotine is high, which accounts for its ability to cross the blood-brain barrier. It is metabolised to cotinine, which shows weak activity toward some nAchR.[29] The maximum response of nicotine is achieved by the administration of a subcutaneous dose of 400 μg/kg.[30]
SYNTHETIC α7-nAchR AGONISTS
A-582941
It is a partial selective α7-nAchR agonist with a high affinity. Its activity toward other nAchR is neglected. It was applied in different studies with variable doses ranging from 0.04 to 10 mg/kg.[31] A-582941 improved negative symptoms and cognition in the ketamine-induced model of schizophrenia in rats.[32] In another study, A-582941 enhanced memory and exhibited sustained pro-cognitive effects after repeated administration.[33] In addition, the administration of A-582941 in combination with other antipsychotic agents was not only useful for improving schizophrenia-associated cognitive defects but it also enhanced the efficacy against positive symptoms and minimised the motor adverse effects associated with these drugs.[31]
A-844606
This drug is derived from tilorone, a known interferon inducer that has a strong selectivity towards α7-nAchR.[34] In a study carried on monkeys and mice, A-844606 showed a high distribution capability in the mouse brain, whereas in monkeys, it was found in higher concentrations in the hippocampus and thalamus than in other brain areas such as the cerebellum. For this reason, it was believed that A-844606 could be a potential ligand for α7-nAchR in the human brain.[35]
AR-R 17779
It is one of the earliest developed selective α7-nAchR agonists.[36] It has a greater affinity for α7-nAchR than other nAchR, including α4β2, α3β2 and α3β4 subtypes.[37] AR-R 17779 is believed to possess cognitive enhancing activity besides its activity in improving social recognition memory in rats.[38,39] Activation of α7-nAchR by AR-R 17779 showed notable anti-inflammatory activity in microglial cells due to the upregulation of α7-nAchR.[40] In addition, AR-R 17779 could be used in the treatment of neurodegenerative diseases such as glaucoma by studying its neuroprotective effect using a purified retinal ganglion cell culture.[41] Moreover, AR-R 17779 activated CAP and ameliorated the post-operative ileus in mice through activation of α7-nAchR.[42]
GTS-21
GTS-21 was initially reported to have cognitive-enhancing activity due to activation of α7-nAchR,[43] then, it was considered a partial selective agonist to α7-nAchR. GTS-21 showed a promising findings in treating dementia when given to healthy male volunteers.[44] In addition, administration of GTS-21 to schizophrenic patients has improved attention and memorisation, which could be related to its ability to modulate the glutamate receptors.[45] The anti-amnesic effect of GTS-21 was investigated in different animal models where it ameliorated cognitive defects in mice injected with β-amyloid by enhancing glutamate activity through an α7-nAchR-mediated mechanism.[46] GTS-21 attenuated cognitive abnormalities and neurodegeneration induced by permanent occlusion of the main carotid arteries, suggesting its beneficial role in the management of neurodegenerative diseases.[47] Moreover, it mitigated the memory impairment induced by isoflurane in aged rats due to its neuroprotective effect.[48] In cerebral ischemia, GTS-21 not only improved memory and learning but also prevented delayed-neuronal death.[49] Furthermore, GTS-21 was investigated in treating nicotine addiction[50] and Alzheimer’s disease.[51] Activation of CAP by GTS-21 attenuated many inflammatory conditions with variant anti-inflammatory mechanisms such as acute renal injury,[52] LPS-induced myocardial injury,[53] burn-induced inflammation[54] and hepatic injury induced by polymicrobial sepsis.[55] In addition, GTS-21 reduced the loss in the body and muscular mass, which may be associated with systemic inflammation[56] in addition to its effect in reducing vascular permeability during endotoxemia.[57]
PHA-543613
It is a novel selective α7-nAchR agonist with negligible or very weak effects on other nAchR.[58] PHA-543613 readily crosses the blood-brain barrier.[59] It was proved that PHA-543613 has neuroprotective activity in neurodegenerative diseases.[59] In Parkinson’s disease model, PHA-543613 protected the damage of dopaminergic neurons through the activation of α7-nAchR.[60] In addition, it improved cognitive impairment and memory deficits associated with schizophrenia.[58,61] PHA-543613 could be useful in treating dementia associated with Alzheimer’s disease, which was proven in β-amyloid-induced amnesic effect in mice.[62,63] Low doses of PHA-543613, at microgram scale, could stimulate the angiogenic process. Its administration in the isoprenaline-induced myocardial infarction model in rats increased capillaries density and restored normal cardiac function and architecture.[64] Moreover, PHA-543613 could attenuate chronic pain related to post-traumatic stress disorder due to the suppression of glial cells.[65] In addition, it reduced neuropathic pain by suppressing dynorphin A activity in microglia.[66]
PNU-282987
It is a potent selective α7-nAchR agonist; however, it showed weak activity on 5-HT3 receptors.[67] Its activity on α7-nAchR has been widely investigated. Centrally, the action of PNU-282987 on spatial learning and cognitive functions is controversial. It showed no effect on the acquisition of spatial learning in the Alzheimer’s disease model.[68] In contrast, another study stated that the administration of PNU-282987 improved cognitive function in mice subjected to β-amyloid, where it reduced apoptosis and increased the level of synaptic associated proteins.[69] Furthermore, it reversed the amnesic effect induced by chlorpheniramine, a histamine receptor antagonist.[70] In phencyclidine-induced cognitive impairments, PNU-282987 daily administration could reverse the learning deficits associated with schizophrenia without causing tolerance.[71] The administration of PNU-282987 could be a promising treatment for Parkinson’s disease. Its daily administration protected astroglia cells, which play a crucial role in maintaining normal brain function. It reduced the apoptosis rate by suppressing anti-apoptotic proteins.[72] In addition, it reduced dopaminergic neurons loss and attenuated neuroinflammation induced by MPTP neurotoxin in mice.[73] PNU-282987 could prevent prolonged febrile seizures by maintaining normal levels of GABA neurotransmitter in the hippocampus.[74] The study of the anti-inflammatory effect of PNU-282987 is promising. The activation of α7-nAchR by PNU-282987 improved the inflammatory profile by modulating macrophages function in acute lung injury.[75] In addition, similar results were obtained in acute pulmonary injury that induced by cardiopulmonary bypass models in rats,[76] sepsis-induced acute lung injury,[77] LPS-induced acute lung injury[78] and acid-induced acute lung injury in rats.[79] In ischemic disorders, PNU-282987 ameliorated cardiac injury due to myocardial reperfusion by modulating beclin-1-mediated autophagy.[80] Furthermore, it showed a protective profile against hepatic injury in the hepatic-reperfusion model by repressing the nuclear factor kappa B (NF-κB) pathway and high-mobility group box 1 protein (HMGB-1) expression.[81] It is thought that PNU-282987 could prevent damage to retinal ganglion cells in glaucoma, which is eventually responsible for blindness that could be due to its ability to stimulate neurogenesis.[82]
Tropisetron
Tropisetron is a potent α7-nAchR agonist with an antagonistic effect on 5-HT3 receptors and α9α10 nAchR subtypes.[83] In ivo administration of tropisetron improved the P50 auditory suppression deficits associated with schizophrenia, suggesting its role as a possible therapeutic target for neurodegenerative diseases.[84] Similar data were obtained by another study in which tropisetron improved the deficient auditory processing in DBA/2 mice due to the activation of α7-nAchR.[85] In addition, the subchronic administration of tropisetron improved cognitive dysfunction raised by the administration of phencyclidine in mice[86] or risperidone-induced schizophrenia.[87] In the ventral tegmental area, tropisetron ameliorated the disruption of dopaminergic neurons of prepulse inhibition and sensorimotor gating deficits.[88] In addition, low doses of tropisetron enhanced memory-related tasks in young or aged rats.[89] Moreover, it could attenuate withdrawal symptoms that occur due to the acute administration of naloxone in rats previously treated with a single dose of morphine, which was explained by its potent effect on α7-nAchR.[90] Tropisetron showed neuroprotective activity by diminishing the excitatory effect of glutamate by stimulating the internalisation of the NMDA glutamate receptor.[91] Tropisetron exerted an α7-nAchR-mediated anti-inflammatory effect in different animal models. In the acetic acid model of ulcerative colitis, it significantly decreased the infiltration of neutrophils and exhibited a notable anti-inflammatory effect, which could be mediated by activation of peroxisome proliferator activated receptor gamma.[92] In addition, tropisetron blocked TNFα-mediated expression of IL-6 and IL-8 in a mechanism independent on p65/NF-κB signaling.[93] Tropisetron attenuated inflammation and acute pancreatitis induced by cerulein, an oligopeptide that stimulates digestive secretions.[94]
ANTAGONISTS OF α7-nAchR
α7-nAchR were developed to discover the mechanism and activity of different nAchR in addition to the generation of impaired nAchR animal models.[95] Some nAchR antagonists were separated from a natural source and many other drugs were chemically synthesised.
α-bungarotoxin
It is a protein toxin that separated from the venom of the Taiwanese banded krait (Bungarus multicinctus).[96] It is a non-selective nAchR antagonist that blocks the muscular nAchR (α1β1γεδ), α7 and α9 nAchR with a great affinity.[96] For this reason, its practical applications are limited to the experimental studies of nAchR activity. A complete blockade is obtained after its preincubation for 1 h at nanomolar concentrations (10 nm), however, the increase in α-bungarotoxin concentration reduces the needed preincubation time.[97] The blockade of nAchR, especially muscular type, is not reversed by washing out due to sow dissociation rate.[98]
Methyllycaconitine (MLA)
It is a reversible nAchR antagonist. It is a norditerpenoid alkaloid separated from the Delphinium species.[99] Its blockade activity is rapid and reversible, making it a suitable alternative to α-bungarotoxin. It has a great binding affinity to α7-nAchR with additional blocking activity to α9 and α9α10 nAchR. However, its binding ability to muscular nAchR is lower.[100] Therefore, MLA is considered a selective α-nAchR antagonist over other nAchR.[101]
Mecamylamine
It is a widely used nAchR antagonist. It was first developed for the treatment of hypertension through blocking the ganglionic receptors.[102] It is highly distributed and crosses the blood-brain barrier, producing a variety of peripheral and central effects.[103] Mecamylamine blocks most neuronal nAchR with more sensitivity toward α3β4 nAchR.[104] The antagonism of α7-nAchR by mecamylamine is reversible.[105] It is used in studying the role of nAchR in behaviour.[95] In addition, the administration of mecamylamine could be efficacious in the treatment of depression.[106]
MOLECULAR MECHANISMS OF CAP
Toll-like receptor 4 (TLR4)/NF-κB
NF-κB plays an important role in regulating the release of TNFα and IL-6 as pro-inflammatory cytokines, which are involved in the inflammatory process. The inflammatory response, which is mediated by NF-κB, is quick as it is previously stored in the cytoplasm in an inactivated form. TLR4 is a pattern recognition receptor, which is activated by different pathogen-associated molecular patterns (PAMPs). This will enhance the innate immune response and initiate inflammatory response.[107] The extracellular part of TLR4 is bound to an intracellular toll-interleukin receptor (TIR) domain that regulates the intracellular signalling cascade of TLR4/MyD88/NF-κB axis, which is ended with formation of pro-inflammatory cytokines.
Many adaptor proteins are essential for the downstream signalling, including myeloid differentiation primary response gene 88 (MyD88), TIR domain-containing adaptor protein (TIRAP) and TIR-domain-containing adaptor-inducing interferon-β. The activation of TLR4/MyD88/NF-κB begins after extracellular activation of TLR4 by PAMPs. The TIRAP is recruited to two TIR domains of two TLR4 where they form a binding site for MyD88.[108] MyD88 forms a complex with two interlein-1 receptor-associated kinases (IRAK2 and IRAK4).[109] After the formation of MyD88/ IRAK complex, TNF receptor-associated factor 6 (TRAF6) is activated forming a trimer [Figure 1]. The IKKγ complex recognises the polyubiquitin chains of TRAF6 leading to its recruitment.[110] This will activate TAK1 and promote the phosphorylation reaction of IκB, which promotes the release and the activation of NF-κB. In addition, TAK1 recruits mitogen-activated protein kinases (MAPKs).[111] Both NF-κB and MAPKs induce the formation and activation of various inflammatory cytokines such as IL-1β, TNF-α and IL-6.[112]
It was previously reported that the translocation of NF-κB and the consequent release of inflammatory cytokines could be prevented by the activation of α7-nAchR [Table 3]. The anti-inflammatory mechanism of α7-nAchR-meditaed blockade of TLR4/NF-κB is not clear and need further investigations.[10] However, it was proven that the α7-nAchR activation suppresses the phosphorylation of Iκ-B by IKK, leading to a consequent suppression of NF-κB production.[8] Furthermore, nicotine was proposed to induce the anti-inflammatory effect of IRAK-M through enhancing its upregulation. The IRAK family consists of two different kinases, active kinases (IRAK-1 and IRAK4) and inactive kinases (IRAK-2 and IRAK-M). IRAK-M serves as a negative regulator of the TLR-4/MyD88 dependent pathway thus, its activation will result in an anti-inflammatory effect.[113] Furthermore, the activation of α7-nAchR by PHA568487 reduced the expression of TLR4, MyD88 and NF-κB in the hippocampus and prevented neurological damage during cardiopulmonary bypass.[114] The same mechanism was obtained by nicotine in airways epithelial cells subjected to LPS.[115] The administration of GTS-21 as a partial selective agonist of α7-nAchR attenuated the Akt/NF-κB pathway in response to LPS challenge.[53,116] In addition, nicotine modulated LPS-induced inflammation and nitric oxide synthesis through suppressing the MAPK pathway.[117] The activation of α7-nAchR alleviated the neurotoxicity and cognitive function induced by β-amyloid protein in the schizophrenic model in mice by inhibiting the MAPK pathway[118] or by blocking PI3K signalling.[119]
| Pathway | Target proteins | Effect of α7-nAchR |
|---|---|---|
| TLR4/NF-κB | Iκ-B | Decreases its phosphorylation by IKK[8] |
| IRAK-M | Induces protein upregulation[113] | |
| TLR4 | Reduces its expression[114] | |
| Myd88 | Reduces its expression[114] | |
| NF-κB | Reduces its expression[114] | |
| Akt | Reduces its activation[53,116] | |
| JAK2/STAT3 | JAK2 | Stimulates its recruitment and autophosphorylation[10] |
| Upregulates its expression[124] | ||
| STAT3 | Stimulates its phosphorylation int pSTAT3[122,123] | |
| pSTAT3 | Stimulates the dimerisation of two molecules of pSTAT3 | |
| TTP | Increases TTP formation, which stimulate CAP[126] | |
| uSTAT | Stimulates its binding to NF-κB forming inactive complex[128] | |
| Nrf2/HO-1 | HO-1 | Increases its activation |
| Increases its expression | ||
| PKC | Enhances its activation by increasing Ca++influx[129] | |
| PI3K/Akt | PI3K | Activation of PI3K, which ends with the activation of Nrf2[133] |
| mTOR | Beclin-1 | Increases its level[141] |
| LCII/I ratio | Increases the ration of ICII/I[141] | |
| NLRP3 | NLRP3 | Decreases its expression[146] |
| Prevents mitochondrial DNA release, which prevent NLRP3 signalling[15] | ||
| β-arrestin-1 | Regulation of its activity where it is essential in the assembly of NLRP3[147] | |
| C-reactive protein | It stimulates α7-nAchR leading to subsequent inhibition of NLRP3.[149,150] | |
| AMPK | p-AMPKα | Increases its expression[152] |
| Cx43 | Enhances the expression of Cx43 by enhancing p-AMPKα[153] | |
| PGC-1α | Enhances mitochondrial biogenesis by stimulating AMPK phosphorylation[155] | |
| NF-κB | Prevents its translocation[156] | |
| AMPK-mTOR | mTOR | Stimulates the autophagy and reduces inflammation |
| HMGB1 | HMGB1 | Restricts its internalisation[162] |
| Prevents its formation[164] | ||
| COX-2 | COX-2 | Increases its expression and suppresses the release of inflammatory cytokines[174,175] |
| PGE2 | Enhances its expression |
Janus activated kinase-2 (JAK2)/signal transducer and activator of transcription 3 (STAT3)
JAK2 is a non-receptor tyrosine kinase, while STAT3 is a member of the STAT family that plays a role as a transcription factor, which is encoded as a STAT3 gene in humans.[120] The JAK2/STAT3 pathway could regulate the CAP.[121] The activation of α7-nAchR results in the recruitment of JAK2 to the α7 subunit, which, in turn, results in autophosphorylation of JAK2 [Table 3].[10] This triggers the phosphorylation process of STAT3 to form phosphorylated STAT3 (pSTAT3). pSTAT3 molecules form dimers that translocate to the nucleus, as shown in [Figure 1]. pSTAT3 dimers act as negative regulators of the inflammatory response.[122,123] Moreover, the activation of α7-nAchR upregulates the expression of JAK2/STAT3 signalling and causes a further suppression in the levels of inflammatory cytokines such as IL-1β.[124]
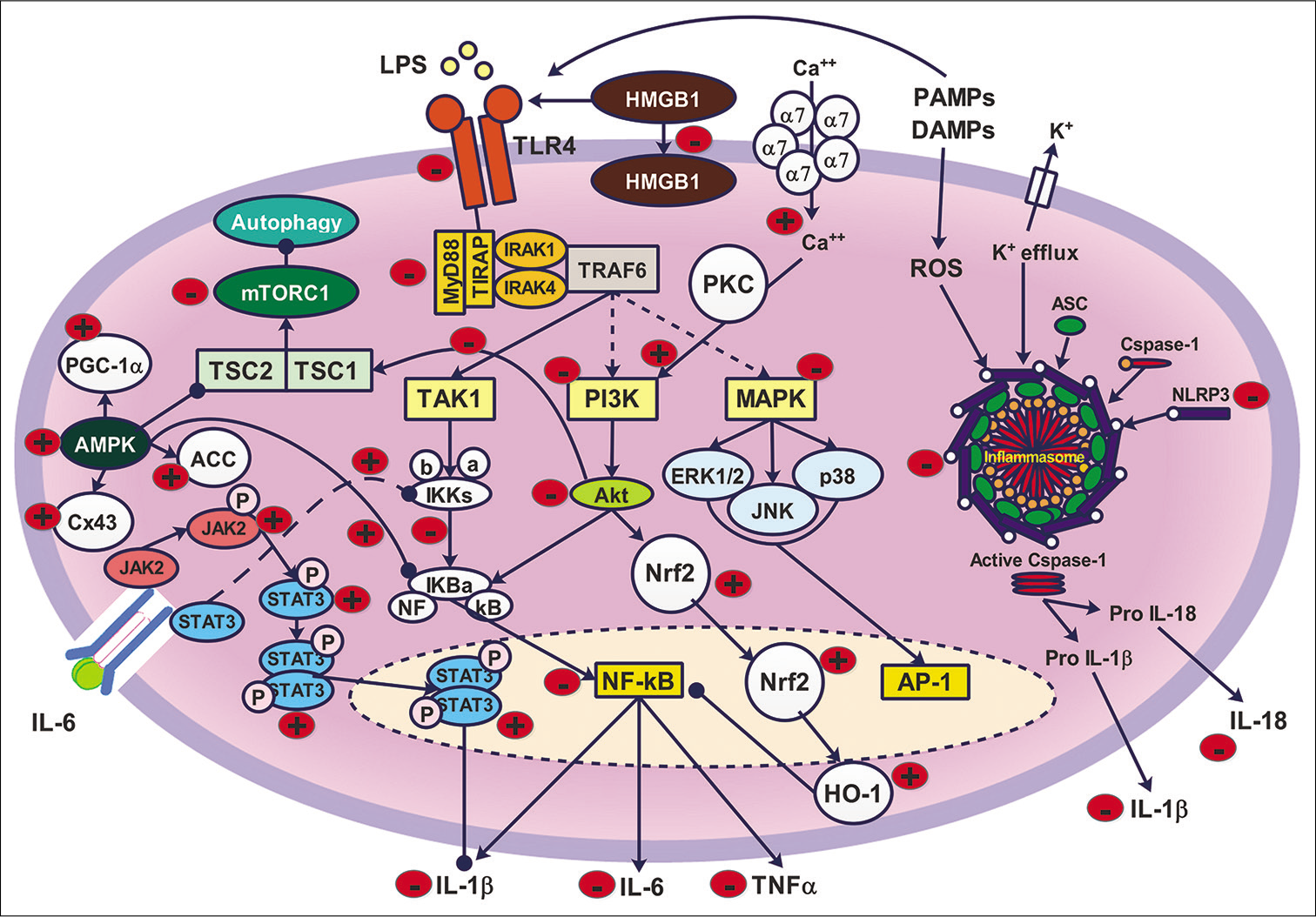
- Schematic diagram of the possible anti-inflammatory mechanism of α7-nAchR. Activation of α7-nAchR inhibits TLR4/NF-κB, where it decreases the expression of TLR4, MyD88, IKKs and NF-κB. In addition, CAP reduced the activity of PI3K and Akt. However, some studies showed that the α7-nAchR could increase PI3K activity, which leads to a consequent increase in Nrf2/HO-1 anti-inflammatory activity. Moreover, activation of α7-nAchR reduces the expression of NLRP3 and reduces the assembly of inflammasome. CAP decreased autophagy and exerted an anti-inflammatory effect by decreasing the activity of mTORC1. It was observed that α7-nAchR stimulation decreased the expression and internalisation of HMGB1. Finally, CAP showed an anti-inflammatory effect through stimulation of anti-inflammatory pathways such as JAK2/STAT3 and AMPK/Cx43/PGC-1α.
In intracerebral haemorrhage model performed in murine models, the administration of PHA-543613 as a selective α7-nAchR agonist attenuated neuroinflammation by activating the JAK2/STAT3 pathway.[125] The mechanism by which the JAK2/STAT3 pathway is regulated by CAP is not completely understood. However, some studies suggested that it is linked to the enhancement of the formation of tristetraprolin (TTP). The enhanced formation of TTP will stimulate the CAP by binding to adenylate-uridylate-rich elements (AU-rich element, AREs).[126] AREs are located in the 3’ untranslated region (3’UTR) of various messenger ribonucleotide (mRNA) molecules that code the transcription of nuclear transcription factors and cytokines[127] In addition, AREs target the rapid degradation of mRNA molecules. Therefore, TTP destabilises the transcripts of pro-inflammatory containing AREs in the 3’UTR. In contrast, another study suggested that unphosphorylated STAT (uSTAT), not pSTAT, plays an essential role in the cholinergic anti-inflammatory effect. uSTAT interferes with the inflammatory response by binding to NF-κB, leading to the displacement of Iκ-B. The formed uSTAT/NF-κB complex could prevent NF-κB activation, giving rise to the α7-nAchR-mediated cholinergic anti-inflammatory effect.[128]
Nuclear factor erythroid 2-related factor 2 (Nrf2)/heme oxygenase-1 (HO-1) induction
CAP is regulated alternatively by activating HO-1 [Figure 1]. Many studies proved that the activation of α7-nAchR could increase the expression of HO-1 and this effect was obliterated when α7-nAchR was blocked selectively by MLA [Table 3]. The possible mechanism of HO-1 activation by α7-nAchR could be due to the enhancement of Ca++ influx. Ca++ ions activate protein kinase C, which, in turn, causes an increase in reactive oxygen species (ROS) through NADPH oxidase-mediated mechanism.[129] This leads to the activation of the PI3K/Akt pathway, which ends with stimulation of Nrf2, leading to the upregulation of HO-1 and the accompanying anti-inflammatory effect. This mechanism was confirmed by studying the effects of PNU-282987 as a selective α7-nAchR agonist in rescuing SHSY5Y cells from apoptosis.[130] In addition, the activation of α7-nAchR protected the kidneys in the acute ischemic renal injury model[131] and endotoxic rats[132] by activating the PI3K/Akt/Nrf2 pathway. In the cerebral ischemia model, the administration of PNU-282987 afforded neuroprotection and anti-inflammatory effects by modulating Nrf2 and HO-1 activity.[133] Moreover, the cholinergic-mediated increase in HO-1 ameliorated inflammatory reactions associated with hepatic ischemia.[134]
Mammalian target of rapamycin (mTOR) signalling
The mTOR participates in internal and external mechanisms that regulate cellular metabolism [Figure 1], growth, proliferation, survival and many other metabolic activities.[135] The mTOR protein belongs to the PI3K family and is differentiated into two distinct multiprotein complexes, mTOR complex 1 and mTOR complex 2 (mTORC1 and mTORC2, respectively).[136] The tuberous sclerosis complex (TSC) is composed of hamartin (TSC1) and tuberin (TSC2) and it is believed to be one of the most important proteins that are involved in the regulation of mTORC1.[137] Inflammation has been shown to regulate mTORC1 signalling, where inflammatory mediators signal mTORC1 through the TSC1/2 complex.[136] The IκB kinase-β is activated by many pro-inflammatory cytokines, such as TNF-α, which plays a role in activating TCS1, leading to activation of mTORC1.[136] It is thought that the positive relationship between mTORC1 and inflammation is important in the development of tumour angiogenesis.[138] Autophagy is defined as an essential metabolic process for the decay of long-lived proteins, damaged tissues and misfolded proteins. It was demonstrated that the autophagy process is very important in the suppression of inflammation in many pathological processes, including myocardial infarction[139] and atherosclerosis.[140] mTOR signalling has an inhibitory effect on autophagy. Many studies showed that the α7-nAchR-mediated anti-inflammatory effect could inhibit the mTOR-related signalling [Table 3]. It was proved that knocking of α7-nAchR aggravated myocardial infarction and the accompanying inflammatory reaction through mTOR-related signalling autophagy. In this study, α7-nAchR deficiency lead to a marked decrease in the autophagy-related proteins such as Beclin-1 and LC3II/I ratio.[141] In addition, the activation of α-nAchR selectively by PNU-282987 significantly alleviated myocardial ischemia through inhibition of the PI3K/mTOR pathway.[80]
Inflammasome
The inflammasome is an intracellular protein complex formed from the nucleotide-binding oligomerisation domain and leucine-rich repeat-containing NOD-like receptors (NLRs) as cytosolic sensors in addition to other proteins, including absent in melanoma 2-like receptors, adapter proteins termed apoptotic speck-containing protein (ASC) or NLR family CARD domain-containing protein 4 and the effector protein pro-caspase-1.[142] The inflammasome assembly [Figure 1] is initiated by recognising molecular patterns such as damage-associated molecular patterns and PAMPs, which are followed by the interaction with ASC. Activated ASC is aggregated into specks, which, in turn, activates procaspase 1 and auto-proteolysis.[143,144] This leads to subsequent cleavage of gasdermin D and elevation of IL-1β and IL-18,[143,144] which will stimulate pyroptosis, which is considered as pro-inflammatory programed cell death.[145] The inflammasome is recognised as a key player in innate immunity. The NLR family, pyrin domain containing 3 (NLRP3) inflammasome connects the innate immunity and the inflammation where it represents one of the most popular inflammatory reactions.[142] It was previously reported that the activation of α7-nAchR could decrease the NLRP3 activity. In the middle cerebral artery occlusion model in rats, the administration of PHA-543613 as a selective α7-nAchR agonist showed a marked inhibitory effect on the NLRP3 activity. This effect was blocked by the administration of α-bungarotoxin, the α7-nAchR antagonist.[146] In another study, it was suggested that activation of α7-nAch mitochondrial receptors could inhibit NLRP3 signalling by preventing mitochondrial DNA release in macrophages.[15] In addition, targeting β-arrestin-1 in monocytes could affect the NLRP3 activation where β-arrestin-1 is essential for its assembly. Therefore, the activation of α7-nAchR could regulates the β-arrestin-1, which leads to a subsequent inhibition of NLRP3 activation.[147] In the animal model of pulmonary hypertension, PNU-282987 administration reduced NLRP3 inflammasome activity and successfully exerted an anti-inflammatory effect and treated pulmonary hypertension.[148] Furthermore, CAP is believed to be modulated endogenously by C-reactive protein, which is formed in the liver and reaches high plasma levels in response to the excessive release of IL-1β in plasma. This could inhibit ATP-induced inflammasome activation due to the stimulatory effect of C-reactive protein on α7-nAchR.[149,150]
Adenosine monophosphate-activated protein kinase (AMPK)
AMPK consists of three subunits, α, β and γ forming a functional enzyme that gives metabolic adaptation by acting as an energy sensor.[151] The adaptation of metabolic energy and the regulation of the immune system exerted by AMPK activation was proven to be protective against many diseases. Activation of CAP could increase the expression of phosphorylated AMPKα, which exerts anti-inflammatory and antifibrotic effects [Figure 1 and Table 3]. In addition, activation of AMPK signalling could suppress the inflammatory response and enhance the expression of gap junction protein connexin 43 (Cx43), which leads to the suppression of IL-1β.[152] Therefore, the preservation of Cx43 could play a critical protective role during inflammatory conditions. AMPK signalling is an important participator in regulating energy balance by several biochemical reactions through the AMPKα/acetyl-CoA carboxylase (ACC) pathway.[153] It is believed that phosphorylation of AMPKα affects the transcriptional cofactor, peroxisome proliferator-activated receptor-γ coactivator (PGC-1α), leading to an enhancement in mitochondrial biogenesis.[154] Activation of CAP by PNU-282987 increases PGC-1α levels through activation of AMPK/ACC signalling.[155] These effects could improve energy metabolism and provide sufficient energy for eukaryotic cells. Moreover, the stimulation of α7-nAchR caused a marked decrease in the nuclear translocation of NF-κB by AMPK-signalling.[156] Furthermore, it was reported that the activation of α7-nAchR in inflammatory bowel disease model in mice stimulate the autophagy and reduce inflammation by the induction of AMPK-mTOR-p70 ribosomal protein S6 kinase pathway. In this study, α7-nAchR-deficient mice showed an injurious effect on bowel inflammation that insulted with dextran sodium sulphate.[157]
HMGB1
HMGB1 is one of the most important chromatin proteins. It interacts with nucleosomes and transcription factors in the nucleus, which regulate DNA transcription.[158] Several immune cells, such as monocytes and macrophages, secrete HMGB1 as an inflammatory mediator cytokine. The mechanism of HMGB1-mediated inflammation is regulated by TLR2 and TLR4 [Figure 1]. The interaction between HMGB1 and TLR4 leads to an elevated expression of NF-κB and MAPK, which leads to a further increase in cytokine levels.[159] In addition, the HMGB1-TLR4 interaction stimulates the formation of ROS by NADPH oxidase.[160] Moreover, the anti-inflammatory effect of HMGB1 could be due to its ability to bind with other-immune activating molecules and facilitate their endocytosis by the aid of the receptor for advanced glycation end-products (RAGE). These pro-inflammatory complexes are targeted to the endolysosomal compartment where HMGB1 permeabilises the lysosomes.[161] It was proved that selective activation of α7-nAchR restricted the internalisation of HMGB1; [Table 3] however, direct inhibition of HMGB1/TLR4 mediated pathway did not suppress the internalisation of HMGB1.[162] The molecular mechanisms of CAP-mediated inhibition of HMGB1 internalisation are still unclear and need further investigation. PNU-282987 prevented the production of HMGB1 and migration in macrophages after LPS administration.[163] Similarly, PNU-282987 gave the same results as it showed a protective effect against hepatic ischemia-reperfusion injury.[81] The lung injury produced by cardiopulmonary bypass was attenuated by electroacupuncture by reducing HMGB1 formation and accompanying inflammatory cytokines by a mechanism that is mediated by α7-nAchR activation.[164] Furthermore, GTS-21 attenuated the production of TNFα, HMGB1 and RAGE in human patients with severe sepsis.[165] In addition, GTS-21 reduced HMGB1 cytoplasmic translocation from macrophages in mice subjected to pulmonary Pseudomonas aeruginosa infection,[166] in addition to its effect on inhibiting the HMGB1/TLR4/NF-κB pathway in radiation-induced lung injury.[167]
Cyclo-oxygenase 2 (COX-2)
COX-2 is the inducible isoform of COX enzymes, which is responsible for the first step in the formation of prostaglandin E2 (PGE2), one of the most important inflammatory mediators that are responsible for several functions of macrophages and lymphocytes.[168] It was shown that PGE2 is associated with many harmful and protective effects, depending on its level. At nanomolar concentrations, PGE2 was proved to exhibit an anti-inflammatory effect in and neuroprotective effect in addition to maintaining cerebral immune hemostasis in a mechanism independent of the PGE2 EP receptors[169] or dependent on EP2 receptors activation.[170] Contrarily, at micromolar concentrations, PGE2 participates in cell death and triggering of apoptosis.[171] It is also involved in the downregulation of microglial cells and the expression of some inflammatory mediators such as TNF-α.[172] Moreover, it was suggested that PGE2 released from macrophages contributes to engulfing apoptotic cell death.[173] It was demonstrated that the selective activation of α7-nAchR enhances the formation of PGE2 by inhibiting the p38/MAPK/NF-κB pathway [Table 3], which could be involved in the alteration of the COX2/PGE2 pathway. This leads to an enhancement in the expression of COX2 enzyme and suppression in the release of pro-inflammatory cytokines.[174,175]
CONCLUSION
The link between the nervous system and the immune system is encouraging to be translated into an effective therapeutic strategy for the treatment of different inflammatory conditions in different body sites. The experimental, pre-clinical and clinical studies showed positive observations. Despite the presence of several α7-nAchR-mediated anti-inflammatory molecular mechanisms, there remain several inflammatory pathways and signalling mechanisms that need further investigation. In addition, more comparative studies are needed to differentiate the anti-inflammatory effects of different α7-nAchR agonists. As these drugs are supposed to be used in the treatment of chronic inflammation, the effect of these drugs on the nervous system should be investigated. In addition, the adverse effects and α7-nAchR independent anti-inflammatory effect should be studied. Finally, the future of using these drugs in the treatment of chronic inflammation is promising and needs more attention.
Declaration of patient consent
Patient's consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Neural regulation of innate immunity: A coordinated nonspecific host response to pathogens. Nat Rev Immunol. 2006;6:318-28.
- [CrossRef] [PubMed] [Google Scholar]
- Transcutaneous auricular vagus nerve stimulation protects endotoxemic rat from lipopolysaccharide-induced inflammation. Evid Based Complement Alternat Med. 2012;2012:627023.
- [CrossRef] [PubMed] [Google Scholar]
- Central muscarinic cholinergic regulation of the systemic inflammatory response during endotoxemia. Proc Natl Acad Sci U S A. 2006;103:5219-23.
- [CrossRef] [PubMed] [Google Scholar]
- Cholinergic modulation of the immune system presents new approaches for treating inflammation. Pharmacol Ther. 2017;179:1-16.
- [CrossRef] [PubMed] [Google Scholar]
- α7-cholinergic receptor mediates vagal induction of splenic norepinephrine. J Immunol. 2011;186:4340-6.
- [CrossRef] [PubMed] [Google Scholar]
- It takes nerve to fight back: The significance of neural innervation of the bone marrow and spleen for immune function. Semin Cell Dev Biol. 2017;61:60-70.
- [CrossRef] [PubMed] [Google Scholar]
- Non-neuronal cholinergic system in regulation of immune function with a focus on α7 nAChRs. Int Immunopharmacol. 2015;29:127-34.
- [CrossRef] [PubMed] [Google Scholar]
- Nicotine inhibits the production of proinflammatory mediators in human monocytes by suppression of I-kappaB phosphorylation and nuclear factor-kappaB transcriptional activity through nicotinic acetylcholine receptor alpha7. Clin Exp Immunol. 2006;146:116-23.
- [CrossRef] [PubMed] [Google Scholar]
- Activation of the α7 nicotinic receptor promotes lipopolysaccharide-induced conversion of M1 microglia to M2. Am J Transl Res. 2017;9:971-85.
- [Google Scholar]
- Activation of the macrophage α7 nicotinic acetylcholine receptor and control of inflammation. J Neuroimmune Pharmacol. 2015;10:468-76.
- [CrossRef] [PubMed] [Google Scholar]
- The structural basis of function in cys-loop receptors. Q Rev Biophys. 2010;43:449-99.
- [CrossRef] [PubMed] [Google Scholar]
- The protective effect of alpha 7 nicotinic acetylcholine receptor activation on critical illness and its mechanism. Int J Biol Sci. 2017;13:46-56.
- [CrossRef] [PubMed] [Google Scholar]
- Structural identification of the nicotinic receptor ion channel. Trends Neurosci. 2018;41:67-70.
- [CrossRef] [PubMed] [Google Scholar]
- α7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol Med. 2014;20:350-8.
- [CrossRef] [PubMed] [Google Scholar]
- Immunohistochemical localisation of nicotinic acetylcholine receptor subunits in human cerebellum. Neuroscience. 2002;113:493-507.
- [CrossRef] [Google Scholar]
- Molecular evolution of the nicotinic acetylcholine receptor: An example of multigene family in excitable cells. J Mol Evol. 1995;40:155-72.
- [CrossRef] [PubMed] [Google Scholar]
- Distribution of α7 nicotinic acetylcholine receptor subunit mRNA in the developing mouse. Front Neuroanat. 2019;13:76.
- [CrossRef] [PubMed] [Google Scholar]
- Nicotinic acetylcholine receptors controlling attention: Behavior, circuits and sensitivity to disruption by nicotine. Biochem Pharmacol. 2013;86:1089-98.
- [CrossRef] [PubMed] [Google Scholar]
- In hippocampal oriens interneurons anti-hebbian long-term potentiation requires cholinergic signaling via α7 nicotinic acetylcholine receptors. J Neurosci. 2013;33:1044-9.
- [CrossRef] [PubMed] [Google Scholar]
- Pharmacokinetic limitations on effects of an alpha7-nicotinic receptor agonist in schizophrenia: Randomized trial with an extended-release formulation. Neuropsychopharmacology. 2018;43:583-9.
- [CrossRef] [PubMed] [Google Scholar]
- Alpha 7 nicotinic acetylcholine receptor and its effects on alzheimer's disease. Neuropeptides. 2019;73:96-106.
- [CrossRef] [PubMed] [Google Scholar]
- Mixed nicotinic-muscarinic properties of the alpha9 nicotinic cholinergic receptor. Neuropharmacology. 2000;39:2515-24.
- [CrossRef] [Google Scholar]
- Alpha7 nicotinic acetylcholine receptor is a target in pharmacology and toxicology. Int J Mol Sci. 2012;13:2219-38.
- [CrossRef] [PubMed] [Google Scholar]
- (+)-Anatoxin-a is a potent agonist at neuronal nicotinic acetylcholine receptors. J Neurochem. 1993;60:2308-11.
- [CrossRef] [PubMed] [Google Scholar]
- Carbamoylcholine homologs: Novel and potent agonists at neuronal nicotinic acetylcholine receptors. Mol Pharmacol. 2003;64:865-75.
- [CrossRef] [PubMed] [Google Scholar]
- High-affinity epibatidine binding of functional, human alpha7-nicotinic acetylcholine receptors stably and heterologously expressed de novo in human SH-EP1 cells. J Pharmacol Exp Ther. 2005;313:24-35.
- [CrossRef] [PubMed] [Google Scholar]
- Nicotine is a selective pharmacological chaperone of acetylcholine receptor number and stoichiometry, Implications for drug discovery. AAPS J. 2009;11:167-77.
- [CrossRef] [PubMed] [Google Scholar]
- The nicotine metabolite, cotinine, alters the assembly and trafficking of a subset of nicotinic acetylcholine receptors. J Biol Chem. 2015;290:24403-12.
- [CrossRef] [PubMed] [Google Scholar]
- Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berl). 2007;190:269-319.
- [CrossRef] [PubMed] [Google Scholar]
- Effects of α7 nicotinic acetylcholine receptor agonists on antipsychotic efficacy in a preclinical mouse model of psychosis. Psychopharmacology (Berl). 2012;220:823-33.
- [CrossRef] [PubMed] [Google Scholar]
- Positive allosteric modulators of alpha 7 nicotinic acetylcholine receptors reverse ketamine-induced schizophrenia-like deficits in rats. Neuropharmacology. 2016;101:389-400.
- [CrossRef] [PubMed] [Google Scholar]
- Differential immediate and sustained memory enhancing effects of alpha7 nicotinic receptor agonists and allosteric modulators in rats. PLoS One. 2011;6:e27014.
- [CrossRef] [PubMed] [Google Scholar]
- α7 nicotinic acetylcholine receptor agonist properties of tilorone and related tricyclic analogues. Br J Pharmacol. 2008;153:1054-61.
- [CrossRef] [PubMed] [Google Scholar]
- In vivo evaluation of α7 nicotinic acetylcholine receptor agonists [11C]A-582941 and [11C]A-844606 in mice and conscious monkeys. PLoS One. 2010;5:e8961.
- [CrossRef] [PubMed] [Google Scholar]
- Selective α7 nicotinic acetylcholine receptor agonists worsen disease in experimental colitis. Br J Pharmacol. 2010;160:322-33.
- [CrossRef] [PubMed] [Google Scholar]
- Activity of alpha7-selective agonists at nicotinic and serotonin 5HT3 receptors expressed in Xenopus oocytes. Bioorg Med Chem Lett. 2004;14:1849-53.
- [CrossRef] [PubMed] [Google Scholar]
- AR-R17779, and alpha7 nicotinic agonist, improves learning and memory in rats. Behav Pharmacol. 1999;10:675-80.
- [CrossRef] [PubMed] [Google Scholar]
- AR-R 17779 improves social recognition in rats by activation of nicotinic alpha7 receptors. Psychopharmacology (Berl). 2004;172:375-83.
- [CrossRef] [PubMed] [Google Scholar]
- Perinatal hypoxiaischemia reduces α 7 nicotinic receptor expression and selective α 7 nicotinic receptor stimulation suppresses inflammation and promotes microglial mox phenotype. Biomed Res Int. 2014;2014:718769.
- [CrossRef] [PubMed] [Google Scholar]
- Characterization and expression of nicotinic ACh receptors in the mammalian retina. Invest Ophthalmol Vis Sci. 2002;43:1.
- [Google Scholar]
- Activation of the cholinergic anti-inflammatory pathway ameliorates postoperative ileus in mice. Gastroenterology. 2007;133:1219-28.
- [CrossRef] [PubMed] [Google Scholar]
- Improved learning and memory in aged rats with chronic administration of the nicotinic receptor agonist GTS-21. Brain Res. 1995;674:252-9.
- [CrossRef] [Google Scholar]
- Safety, pharmacokinetics, and effects on cognitive function of multiple doses of GTS-21 in healthy, male volunteers. Neuropsychopharmacology. 2003;28:542-51.
- [CrossRef] [PubMed] [Google Scholar]
- Effects of the nicotinic α7 receptor partial agonist GTS-21 on NMDA-glutamatergic receptor related deficits in sensorimotor gating and recognition memory in rats. Psychopharmacology (Berl). 2014;231:3695-706.
- [CrossRef] [PubMed] [Google Scholar]
- DMXB (GTS-21) ameliorates the cognitive deficits in beta amyloid(25-35(-)) injected mice through preventing the dysfunction of alpha7 nicotinic receptor. J Neurosci Res. 2010;88:1784-94.
- [CrossRef] [PubMed] [Google Scholar]
- GTS-21, a nicotinic agonist, attenuates multiple infarctions and cognitive deficit caused by permanent occlusion of bilateral common carotid arteries in rats. Jpn J Pharmacol. 1998;78:463-9.
- [CrossRef] [PubMed] [Google Scholar]
- Alpha 7 nicotinic acetylcholine receptor agonist GTS-21 mitigates isoflurane-induced cognitive impairment in aged rats. J Surg Res. 2015;194:255-61.
- [CrossRef] [PubMed] [Google Scholar]
- Protective effect of GTS-21, a novel nicotinic receptor agonist, on delayed neuronal death induced by ischemia in gerbils. Jpn J Pharmacol. 1998;76:23-9.
- [CrossRef] [PubMed] [Google Scholar]
- Advances in pharmacotherapy for tobacco dependence. Expert Opin Emerg Drugs. 2004;9:39-53.
- [CrossRef] [PubMed] [Google Scholar]
- Alpha7 nicotinic acetylcholine receptor-specific agonist DMXBA (GTS-21) attenuates Aβ accumulation through suppression of neuronal γ-secretase activity and promotion of microglial amyloid-β phagocytosis and ameliorates cognitive impairment in a mouse model of alzheimer's disease. Neurobiol Aging. 2018;62:197-209.
- [CrossRef] [PubMed] [Google Scholar]
- Activation of the cholinergic anti-inflammatory pathway by GTS-21 attenuates cisplatin-induced acute kidney injury in mice. PLoS One. 2017;12:e0188797.
- [CrossRef] [PubMed] [Google Scholar]
- GTS-21 protected against LPS-induced sepsis myocardial injury in mice through α7nAChR. Inflammation. 2018;41:1073-83.
- [CrossRef] [PubMed] [Google Scholar]
- Prevention of burn-induced inflammatory responses and muscle wasting by GTS-21, a specific agonist for α7 nicotinic acetylcholine receptors. Shock. 2017;47:61-9.
- [CrossRef] [PubMed] [Google Scholar]
- GTS-21 ameliorates polymicrobial sepsis-induced hepatic injury by modulating autophagy through α7nAchRs in mice. Cytokine. 2020;128:155019.
- [CrossRef] [PubMed] [Google Scholar]
- GTS-21 attenuates loss of body mass, muscle mass, and function in rats having systemic inflammation with and without disuse atrophy. Pflugers Arch. 2018;470:1647-57.
- [CrossRef] [PubMed] [Google Scholar]
- GTS-21 reduces microvascular permeability during experimental endotoxemia. Microvasc Res. 2018;115:75-82.
- [CrossRef] [PubMed] [Google Scholar]
- Discovery of N-[(3R)-1-azabicyclo [2.2.2]oct-3-yl] furo [2,3-c] pyridine-5-carboxamide, an agonist of the alpha7 nicotinic acetylcholine receptor, for the potential treatment of cognitive deficits in schizophrenia: Synthesis and structure-activity relationship. J Med Chem. 2006;49:4425-36.
- [CrossRef] [PubMed] [Google Scholar]
- Neuroprotective effect of the alpha 7 nicotinic receptor agonist PHA 543613 in an in vivo excitotoxic adult rat model. Neuroscience. 2017;356:52-63.
- [CrossRef] [PubMed] [Google Scholar]
- Assessment of the protection of dopaminergic neurons by an α7 nicotinic receptor agonist, PHA 543613 using [(18)F] LBT-999 in a parkinson's disease rat model. Front Med (Lausanne). 2015;2:61.
- [CrossRef] [PubMed] [Google Scholar]
- Effects of haloperidol, olanzapine, ziprasidone, and PHA-543613 on spatial learning and memory in the Morris water maze test in naive and MK-801-treated mice. Brain Behav. 2017;7:e00764.
- [CrossRef] [PubMed] [Google Scholar]
- Effect of alpha-7 nicotinic acetylcholine receptor activation on beta-amyloid induced recognition memory impairment, Possible role of neurovascular function. Acta Cir Bras. 2015;30:736-42.
- [CrossRef] [PubMed] [Google Scholar]
- Selective activation of α7 nicotinic acetylcholine receptor by PHA-543613 improves Aβ25-35-mediated cognitive deficits in mice. Neuroscience. 2015;298:81-93.
- [CrossRef] [PubMed] [Google Scholar]
- α7-nAChR-mediated therapeutic angiogenesis accounts for the advantageous effect of low nicotine doses against myocardial infarction in rats. FASEB J. 2019;33:679.1.
- [Google Scholar]
- Spinal activation of alpha7-nicotinic acetylcholine receptor attenuates posttraumatic stress disorder-related chronic pain via suppression of glial activation. Neuroscience. 2017;344:243-54.
- [CrossRef] [PubMed] [Google Scholar]
- Activation of alpha7 acetylcholine receptors reduces neuropathic pain by decreasing dynorphin A release from microglia. Brain Res. 2019;1715:57-65.
- [CrossRef] [PubMed] [Google Scholar]
- The selective alpha7 nicotinic acetylcholine receptor agonist PNU-282987 [N-[(3R)-1-Azabicyclo [2.2.2] oct-3-yl]-4-chlorobenzamide hydrochloride] enhances GABAergic synaptic activity in brain slices and restores auditory gating deficits in anesthetized rats. J Pharmacol Exp Ther. 2005;312:1213-22.
- [CrossRef] [PubMed] [Google Scholar]
- Behavioural effects of PNU-282987 and stress in an animal model of alzheimer's disease. Psychogeriatrics. 2017;17:33-42.
- [CrossRef] [PubMed] [Google Scholar]
- Activation of α7 nAChR by PNU-282987 improves synaptic and cognitive functions through restoring the expression of synaptic-associated proteins and the CaM-CaMKII-CREB signaling pathway. Aging (Albany NY). 2020;12:543-70.
- [CrossRef] [PubMed] [Google Scholar]
- Cholinergic agonist reverses H1-induced memory deficit in mice. Prog Neuropsychopharmacol Biol Psychiatry. 2017;72:16-22.
- [CrossRef] [PubMed] [Google Scholar]
- Activation of α7 nicotinic receptors improves phencyclidine-induced deficits in cognitive tasks in rats: Implications for therapy of cognitive dysfunction in schizophrenia. Eur Neuropsychopharmacol. 2011;21:333-43.
- [CrossRef] [PubMed] [Google Scholar]
- Activation of α7 nicotinic acetylcholine receptor protects against 1-methyl-4-phenylpyridinium-induced astroglial apoptosis. Front Cell Neurosci. 2019;13:507.
- [CrossRef] [PubMed] [Google Scholar]
- The α7 nAChR agonist PNU-282987 reduces inflammation and MPTP-induced nigral dopaminergic cell loss in mice. J Parkinsons Dis. 2013;3:161-72.
- [CrossRef] [PubMed] [Google Scholar]
- Acetylcholine receptor agonist effect on seizure activity and GABAergic mechanisms involved in prolonged febrile seizure development in an animal model. Brain Res Bull. 2019;149:203-7.
- [CrossRef] [PubMed] [Google Scholar]
- Acute lung injury is reduced by the α7nAChR agonist PNU-282987 through changes in the macrophage profile. FASEB J. 2017;31:320-32.
- [CrossRef] [PubMed] [Google Scholar]
- Alpha7 Nicotine acetylcholine receptor agonist PNU-282987 attenuates acute lung injury in a cardiopulmonary bypass model in rats. Shock. 2017;47:474-9.
- [CrossRef] [PubMed] [Google Scholar]
- Protective effects of PNU-282987 on sepsis-induced acute lung injury in mice. Mol Med Rep. 2019;19:3791-8.
- [CrossRef] [PubMed] [Google Scholar]
- Activation of alpha-7 nicotinic acetylcholine receptors (α7nAchR) promotes the protective autophagy in LPS-induced acute lung injury (ALI) in vitro and in vivo. Inflammation. 2019;42:2236-45.
- [CrossRef] [PubMed] [Google Scholar]
- Activation of the α7 nAChR reduces acid-induced acute lung injury in mice and rats. Am J Respir Cell Mol Biol. 2007;37:186-92.
- [CrossRef] [PubMed] [Google Scholar]
- Alpha7 nicotinic acetylcholine receptor activation protects against myocardial reperfusion injury through modulation of autophagy. Biochem Biophys Res Commun. 2018;500:357-64.
- [CrossRef] [PubMed] [Google Scholar]
- The protective effect of PNU-282987, a selective α7 nicotinic acetylcholine receptor agonist, on the hepatic ischemia-reperfusion injury is associated with the inhibition of high-mobility group box 1 protein expression and nuclear factor κB activation in mice. Shock. 2013;39:197-203.
- [CrossRef] [PubMed] [Google Scholar]
- Evidence of BrdU positive retinal neurons after application of an alpha7 nicotinic acetylcholine receptor agonist. Neuroscience. 2017;346:437-46.
- [CrossRef] [PubMed] [Google Scholar]
- Occupancy of α7 nicotinic acetylcholine receptors in the brain by tropisetron: A positron emission tomography study using [(11)C] CHIBA-1001 in healthy human subjects. Clin Psychopharmacol Neurosci. 2011;9:111-6.
- [CrossRef] [PubMed] [Google Scholar]
- Tropisetron improves deficits in auditory P50 suppression in schizophrenia. Schizophr Res. 2005;76:67-72.
- [CrossRef] [PubMed] [Google Scholar]
- Tropisetron improves deficient inhibitory auditory processing in DBA/2 mice: Role of alpha 7 nicotinic acetylcholine receptors. Psychopharmacology (Berl). 2005;183:13-9.
- [CrossRef] [PubMed] [Google Scholar]
- Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of tropisetron: Role of alpha7 nicotinic receptors. Eur J Pharmacol. 2006;553:191-5.
- [CrossRef] [PubMed] [Google Scholar]
- Tropisetron enhances recognition memory in rats chronically treated with risperidone or quetiapine. Biochem Pharmacol. 2018;151:180-7.
- [CrossRef] [PubMed] [Google Scholar]
- Ameliorating effects of tropisetron on dopaminergic disruption of prepulse inhibition via the alpha(7) nicotinic acetylcholine receptor in wistar rats. Brain Res. 2010;1353:152-8.
- [CrossRef] [PubMed] [Google Scholar]
- Tropisetron sensitizes α7 containing nicotinic receptors to low levels of acetylcholine in vitro and improves memory-related task performance in young and aged animals. Neuropharmacology. 2017;117:422-33.
- [CrossRef] [PubMed] [Google Scholar]
- Tropisetron attenuates naloxone-induced place aversion in single-dose morphine-treated rats: Role of alpha7 nicotinic receptors. Eur J Pharmacol. 2009;609:74-7.
- [CrossRef] [PubMed] [Google Scholar]
- Tropisetron as a neuroprotective agent against glutamate-induced excitotoxicity and mechanisms of action. Neuropharmacology. 2013;73:111-21.
- [CrossRef] [PubMed] [Google Scholar]
- Involvement of PPARγ in the protective action of tropisetron in an experimental model of ulcerative colitis. Immunopharmacol Immunotoxicol. 2016;38:432-40.
- [CrossRef] [PubMed] [Google Scholar]
- Tropisetron via α7 nicotinic acetylcholine receptor suppresses tumor necrosis factor-α−mediated cell responses of human keratinocytes. Exp Dermatol. 2019;28:276-82.
- [CrossRef] [PubMed] [Google Scholar]
- Protective effects of tropisetron on cerulein-induced acute pancreatitis in mice. Biomed Pharmacother. 2017;93:589-95.
- [CrossRef] [PubMed] [Google Scholar]
- Nicotinic receptor antagonists in rats In: Levin ED, Buccafusco JJ, eds. Animal Models of Cognitive Impairment. Boca Raton, FL: CRC Press, Taylor & Francis; 2006.
- [Google Scholar]
- The nicotinic acetylcholine receptor: The founding father of the pentameric ligand-gated ion channel superfamily. J Biol Chem. 2012;287:40207-15.
- [CrossRef] [PubMed] [Google Scholar]
- Nootropic alpha7 nicotinic receptor allosteric modulator derived from GABAA receptor modulators. Proc Natl Acad Sci U S A. 2007;104:8059-64.
- [CrossRef] [PubMed] [Google Scholar]
- Neuronal control of the cardiac responses to osmotic stress in the gastropod limpet Patella caerulea. J Exp Zool A Comp Exp Biol. 2006;305:472-9.
- [CrossRef] [PubMed] [Google Scholar]
- Methyllycaconitine: A selective probe for neuronal alpha-bungarotoxin binding sites. FEBS Lett. 1990;270:45-8.
- [CrossRef] [Google Scholar]
- Expression patterns of nicotinic subunits α2, α7, α8, and β1 affect the kinetics and pharmacology of ACh-induced currents in adult bee olfactory neuropiles. J Neurophysiol. 2011;106:1604-13.
- [CrossRef] [PubMed] [Google Scholar]
- The α3β4* nicotinic acetylcholine receptor subtype mediates nicotine reward and physical nicotine withdrawal signs independently of the α5 subunit in the mouse. Neuropharmacology. 2013;70:228-35.
- [CrossRef] [PubMed] [Google Scholar]
- Nicotinic acetylcholine receptors: From basic science to therapeutics. Pharmacol Ther. 2013;137:22-54.
- [CrossRef] [PubMed] [Google Scholar]
- Similar activity of mecamylamine stereoisomers in vitro and in vivo. Eur J Pharmacol. 2013;720:264-75.
- [CrossRef] [PubMed] [Google Scholar]
- Mecamylamine-a nicotinic acetylcholine receptor antagonist with potential for the treatment of neuropsychiatric disorders. Expert Opin Pharmacother. 2009;10:2709-21.
- [CrossRef] [PubMed] [Google Scholar]
- Behavioral effects of nicotinic antagonist mecamylamine in a rat model of depression: Prefrontal cortex level of BDNF protein and monoaminergic neurotransmitters. Psychopharmacology (Berl). 2015;232:1095-105.
- [CrossRef] [PubMed] [Google Scholar]
- Cardioprotective role of GTS-21 by attenuating the TLR4/NF-κB pathway in streptozotocin-induced diabetic cardiomyopathy in rats. Naunyn Schmiedebergs Arch Pharmacol 2020
- [CrossRef] [PubMed] [Google Scholar]
- Functional interfaces between TICAM-2/TRAM and TICAM-1/TRIF in TLR4 signaling. Biochem Soc Trans. 2017;45:929-35.
- [CrossRef] [PubMed] [Google Scholar]
- Interleukin-1 receptor-associated kinase 4 (IRAK4) plays a dual role in myddosome formation and Toll-like receptor signaling. J Biol Chem. 2018;293:15195-207.
- [CrossRef] [PubMed] [Google Scholar]
- IRAK4 dimerization and trans-autophosphorylation are induced by myddosome assembly. Mol Cell. 2014;55:891-903.
- [CrossRef] [PubMed] [Google Scholar]
- Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50-83.
- [CrossRef] [PubMed] [Google Scholar]
- Angiotensin II: A key mediator in the development of liver fibrosis and cancer. Bull Natl Res Cent. 2018;42:18.
- [CrossRef] [Google Scholar]
- A new IRAK-M-mediated mechanism implicated in the anti-inflammatory effect of nicotine via α7 nicotinic receptors in human macrophages. PLoS One. 2014;9:e108397.
- [CrossRef] [PubMed] [Google Scholar]
- α7 nicotinic acetylcholine receptor agonist inhibits the damage of rat hippocampal neurons by TLR4/Myd88/NF-κB signaling pathway during cardiopulmonary bypass. Mol Med Rep. 2017;16:4770-6.
- [CrossRef] [PubMed] [Google Scholar]
- Nicotine reduces TNF-α expression through a α7 nAChR/MyD88/NF-ĸB pathway in HBE16 airway epithelial cells. Cell Physiol Biochem. 2011;27:605-12.
- [CrossRef] [PubMed] [Google Scholar]
- GTS-21 attenuates lipopolysaccharide-induced inflammatory cytokine production in vitro by modulating the Akt and NF-κB signaling pathway through the α7 nicotinic acetylcholine receptor. Int Immunopharmacol. 2015;29:504-12.
- [CrossRef] [PubMed] [Google Scholar]
- Inhibition of lipopolysaccharide-induced nitric oxide synthesis by nicotine through S6K1-p42/44 MAPK pathway and STAT3 (Ser 727) phosphorylation in raw 264.7 cells. Cytokine. 2008;44:126-34.
- [CrossRef] [PubMed] [Google Scholar]
- Activation of α7 nicotinic acetylcholine receptor alleviates Aβ1-42-induced neurotoxicity via downregulation of p38 and JNK MAPK signaling pathways. Neurochem Int. 2018;120:238-50.
- [CrossRef] [PubMed] [Google Scholar]
- Alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J Biol Chem. 2001;276:13541-6.
- [CrossRef] [PubMed] [Google Scholar]
- Do we know jack About JAK? A closer look at JAK/STAT signaling pathway. Front Oncol. 2018;8:287.
- [CrossRef] [PubMed] [Google Scholar]
- α7 nicotinic acetylcholine receptor mediates the neuroprotection of remote ischemic postconditioning in a rat model of asphyxial cardiac arrest. J Surg Res. 2020;246:6-18.
- [CrossRef] [PubMed] [Google Scholar]
- JAK2-STAT3 signaling: A novel function and a novel mechanism. JAKSTAT. 2012;1:191-3.
- [CrossRef] [PubMed] [Google Scholar]
- Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6:844-51.
- [CrossRef] [PubMed] [Google Scholar]
- Gx-50 inhibits neuroinflammation via α7 nAChR activation of the JAK2/STAT3 and PI3K/AKT pathways. J Alzheimers Dis. 2016;50:859-71.
- [CrossRef] [PubMed] [Google Scholar]
- α 7 nicotinic acetylcholine receptor stimulation attenuates neuroinflammation through JAK2-STAT3 activation in murine models of intracerebral hemorrhage. Biomed Res Int. 2017;2017:8134653.
- [CrossRef] [PubMed] [Google Scholar]
- Inhibitory effects of nicotine derived from cigarette smoke on thymic stromal lymphopoietin production in epidermal keratinocytes. Cell Immunol. 2016;302:19-25.
- [CrossRef] [PubMed] [Google Scholar]
- Lessons from studying the AU-rich elements in chronic inflammation and autoimmunity. J Autoimmun. 2019;104:102334.
- [CrossRef] [PubMed] [Google Scholar]
- Unphosphorylated STAT3 modulates alpha 7 nicotinic receptor signaling and cytokine production in sepsis. Eur J Immunol. 2010;40:2580-9.
- [CrossRef] [PubMed] [Google Scholar]
- Modulatory effects of α7 nAChRs on the immune system and its relevance for CNS disorders. Cell Mol Life Sci. 2016;73:2511-30.
- [CrossRef] [PubMed] [Google Scholar]
- Poststress treatment with PNU282987 can rescue SH-SY5Y cells undergoing apoptosis via α7 nicotinic receptors linked to a Jak2/Akt/HO-1 signaling pathway. Free Radic Biol Med. 2010;49:1815-21.
- [CrossRef] [PubMed] [Google Scholar]
- The proximal tubular α7 nicotinic acetylcholine receptor attenuates ischemic acute kidney injury through Akt/PKC signaling-mediated HO-1 induction. Exp Mol Med. 2018;50:40.
- [CrossRef] [PubMed] [Google Scholar]
- The α7-nAChR/ heme oxygenase-1/carbon monoxide pathway mediates the nicotine counteraction of renal inflammation and vasoconstrictor hyporeactivity in endotoxic male rats. Inflamm Res. 2020;69:217-31.
- [CrossRef] [PubMed] [Google Scholar]
- The microglial α7-acetylcholine nicotinic receptor is a key element in promoting neuroprotection by inducing heme oxygenase-1 via nuclear factor erythroid-2-related factor 2. Antioxid Redox Signal. 2013;19:1135-48.
- [CrossRef] [PubMed] [Google Scholar]
- Activation of the cholinergic anti-inflammatory pathway by nicotine attenuates hepatic ischemia/reperfusion injury via heme oxygenase-1 induction. Eur J Pharmacol. 2013;707:61-70.
- [CrossRef] [PubMed] [Google Scholar]
- Mammalian target of rapamycin: Discovery of rapamycin reveals a signaling pathway important for normal and cancer cell growth. Semin Oncol. 2009;36(Suppl 3):S3-17.
- [CrossRef] [PubMed] [Google Scholar]
- Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv Biol Regul. 2019;72:51-62.
- [CrossRef] [PubMed] [Google Scholar]
- Tuberous sclerosis complex: Review based on new diagnostic criteria. An Bras Dermatol. 2018;93:323-31.
- [CrossRef] [Google Scholar]
- PGE2-induced colon cancer growth is mediated by mTORC1. Biochem Biophys Res Commun. 2014;451:587-91.
- [CrossRef] [PubMed] [Google Scholar]
- The role of autophagy in acute myocardial infarction. Front Pharmacol. 2019;10:551.
- [CrossRef] [PubMed] [Google Scholar]
- Role of autophagy in atherosclerosis: Foe or friend? J Inflamm (Lond). 2019;16:8.
- [CrossRef] [PubMed] [Google Scholar]
- α7nAChR deletion aggravates myocardial infarction and enhances systemic inflammatory reaction via mTOR-signaling-related autophagy. Inflammation. 2019;42:1190-202.
- [CrossRef] [PubMed] [Google Scholar]
- Metabolic regulation of inflammasomes in inflammation. Immunology. 2019;157:95-109.
- [CrossRef] [PubMed] [Google Scholar]
- Critical role of apoptotic speck protein containing a caspase recruitment domain (ASC) and NLRP3 in causing necrosis and ASC speck formation induced by Porphyromonas gingivalis in human cells. J Immunol. 2009;182:2395-404.
- [CrossRef] [PubMed] [Google Scholar]
- Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe. 2010;8:471-83.
- [CrossRef] [PubMed] [Google Scholar]
- Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660-5.
- [CrossRef] [PubMed] [Google Scholar]
- Electroacupuncture attenuated cerebral ischemic injury and neuroinflammation through α7nAChR-mediated inhibition of NLRP3 inflammasome in stroke rats. Mol Med. 2019;25:22.
- [CrossRef] [PubMed] [Google Scholar]
- Activating α7 nicotinic acetylcholine receptor inhibits NLRP3 inflammasome through regulation of β-arrestin-1. CNS Neurosci Ther. 2017;23:875-84.
- [CrossRef] [PubMed] [Google Scholar]
- Activation of nicotinic acetylcholine α7 receptor attenuates progression of monocrotaline-induced pulmonary hypertension in rats by downregulating the NLRP3 inflammasome. Front Pharmacol. 2019;10:128.
- [CrossRef] [PubMed] [Google Scholar]
- Canonical and novel non-canonical cholinergic agonists inhibit ATP-induced release of monocytic interleukin-1β via different combinations of nicotinic acetylcholine receptor subunits α7, α9 and α10. Front Cell Neurosci. 2017;11:189.
- [CrossRef] [PubMed] [Google Scholar]
- C-reactive protein stimulates nicotinic acetylcholine receptors to control ATP-mediated monocytic inflammasome activation. Front Immunol. 2018;9:1604.
- [CrossRef] [PubMed] [Google Scholar]
- AMPK: Regulation of metabolic dynamics in the context of autophagy. Int J Mol Sci. 2018;19:3812.
- [CrossRef] [PubMed] [Google Scholar]
- Inhibition of adenosine monophosphate-activated protein kinase reduces glial cell-mediated inflammation and induces the expression of Cx43 in astroglias after cerebral ischemia. Brain Res. 2015;1605:1-11.
- [CrossRef] [PubMed] [Google Scholar]
- AMPK signaling to acetyl-CoA carboxylase is required for fasting-and cold-induced appetite but not thermogenesis. Elife. 2018;7:e32656.
- [CrossRef] [PubMed] [Google Scholar]
- PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014;16:992-1003, 1-15
- [CrossRef] [PubMed] [Google Scholar]
- Heme-oxygenase I and PCG-1α regulate mitochondrial biogenesis via microglial activation of alpha7 nicotinic acetylcholine receptors using PNU282987. Antioxid Redox Signal. 2017;27:93-105.
- [CrossRef] [PubMed] [Google Scholar]
- Eliciting α7-nAChR exerts cardioprotective effects on ischemic cardiomyopathy via activation of AMPK signalling. J Cell Mol Med. 2019;23:4746-58.
- [CrossRef] [PubMed] [Google Scholar]
- Alpha7 nicotinic acetylcholine receptor alleviates inflammatory bowel disease through induction of AMPK-mTOR-p70S6K-mediated autophagy. Inflammation. 2019;42:1666-79.
- [CrossRef] [PubMed] [Google Scholar]
- HMGB proteins: Interactions with DNA and chromatin. Biochim Biophys Acta. 2010;1799:101-13.
- [CrossRef] [PubMed] [Google Scholar]
- Huang Qi tong Bi decoction attenuates myocardial ischemia-reperfusion injury via HMGB1/TLR/NF-κB pathway. Mediators Inflamm. 2019;2019:8387636.
- [CrossRef] [PubMed] [Google Scholar]
- Myeloid differentiation protein 2 induced retinal ischemia reperfusion injury via upregulation of ROS through a TLR4-NOX4 pathway. Toxicol Lett. 2018;282:109-20.
- [CrossRef] [PubMed] [Google Scholar]
- High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin Immunol. 2018;38:40-8.
- [CrossRef] [PubMed] [Google Scholar]
- Inhibition of HMGB1/RAGE-mediated endocytosis by HMGB1 antagonist box A, anti-HMGB1 antibodies, and cholinergic agonists suppresses inflammation. Mol Med. 2019;25:13.
- [CrossRef] [PubMed] [Google Scholar]
- Requisite role of the cholinergic α7 nicotinic acetylcholine receptor pathway in suppressing gram-negative sepsis-induced acute lung inflammatory injury. J Immunol. 2010;184:401-10.
- [CrossRef] [PubMed] [Google Scholar]
- Electroacupuncture pretreatment attenuates acute lung injury through α7 nicotinic acetylcholine receptor-mediated inhibition of HMGB1 release in rats after cardiopulmonary bypass. Shock. 2018;50:351-9.
- [CrossRef] [PubMed] [Google Scholar]
- The selective α7 agonist GTS-21 attenuates cytokine production in human whole blood and human monocytes activated by ligands for TLR2, TLR3, TLR4, TLR9, and RAGE. Mol Med. 2009;15:195-202.
- [CrossRef] [PubMed] [Google Scholar]
- The α7 nicotinic acetylcholine receptor agonist GTS-21 improves bacterial clearance in mice by restoring hyperoxia-compromised macrophage function. Mol Med. 2014;20:238-47.
- [CrossRef] [PubMed] [Google Scholar]
- α7-nAchR agonist GTS-21 reduces radiation-induced lung injury. Oncol Rep. 2018;40:2287-97.
- [CrossRef] [PubMed] [Google Scholar]
- Reactive astrocyte COX2-PGE2 production inhibits oligodendrocyte maturation in neonatal white matter injury. Glia. 2017;65:2024-37.
- [CrossRef] [PubMed] [Google Scholar]
- Physiological concentration of prostaglandin E2 exerts anti-inflammatory effects by inhibiting microglial production of superoxide through a novel pathway. Mol Neurobiol. 2018;55:8001-13.
- [CrossRef] [PubMed] [Google Scholar]
- Stimulation of PGE receptors EP2 and EP4 protects cultured neurons against oxidative stress and cell death following beta-amyloid exposure. Eur J Neurosci. 2005;22:2199-206.
- [CrossRef] [PubMed] [Google Scholar]
- PGE2-induced apoptotic cell death in K562 human leukaemia cells. Cell Physiol Biochem. 2006;17:201-10.
- [CrossRef] [PubMed] [Google Scholar]
- Inducible nitric oxide synthase expression in activated rat microglial cultures is downregulated by exogenous prostaglandin E2 and by cyclooxygenase inhibitors. Glia. 1997;19:152-60.
- [CrossRef] [Google Scholar]
- Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890-8.
- [CrossRef] [PubMed] [Google Scholar]
- Microsomal prostaglandin E synthase-1 gene deletion impairs neuro-immune circuitry of the cholinergic anti-inflammatory pathway in endotoxaemic mouse spleen. PLoS One. 2018;13:e0193210.
- [CrossRef] [PubMed] [Google Scholar]
- Activation of α7 nicotinic acetylcholine receptor by nicotine selectively up-regulates cyclooxygenase-2 and prostaglandin E2 in rat microglial cultures. J Neuroinflammation. 2005;2:4.
- [CrossRef] [PubMed] [Google Scholar]




